Advanced pharmaceutical bulletin. 11(2):343-350.
doi: 10.34172/apb.2021.032
Research Article
Multiplex Genome Editing in Chinese Hamster Ovary Cell Line Using All-in-One and HITI CRISPR Technology
Fatemeh Safari 1, 2  , Safar Farajnia 3, * , Younes Ghasemi 4, Nosratollah Zarghami 1, Mazyar Barekati Mowahed 5
, Safar Farajnia 3, * , Younes Ghasemi 4, Nosratollah Zarghami 1, Mazyar Barekati Mowahed 5
Author information:
1Medical Biotechnology Department, Faculty of Advanced Medical Sciences, Tabriz University of Medical Sciences, Tabriz, Iran.
2Student Research Committee, Tabriz University of Medical Sciences, Tabriz, Iran.
3Biotechnology Research Center, Tabriz University of Medical Sciences, Tabriz, Iran.
4Department of Pharmaceutical Biotechnology, School of Pharmacy, and Pharmaceutical Sciences Research Centre, Shiraz University of Medical Sciences, Shiraz, Iran.
5Department of Physiology & Biophysics, School of Medicine, Case Western Reserve University, Ohio, USA.
Abstract
Purpose:
CRISPR/Cas9 gene editing technology has revolutionized gene manipulation by providing the opportunity of gene knock out/in, transcriptional modification and base editing. The application of this system extended into different eras of biology, from cell development to animal modeling. Various generations of CRISPR technology have been developed to make genome editing easy which resulted in rapid protocols for amelioration of a large genome.
Methods: We established a simple protocol for gene manipulation in Chinese hamster ovary (CHO) cells to achieve a Caspase 7 deficient cell line by using combination of all-in-one CRISPR technology and CRISPR/Cas9 homology-independent targeted integration (CRISPR HITI).
Results: the findings of this study indicated that using CRISPR knocking in/out technology facilitates genomic manipulation in CHO cells. Integration of EGFP in target locus of caspase 7 gene made the selection of knockout CHO cell line easy which achieved by cell sorting and single-cell cloning.
Conclusion: this system introduces an effective targeting strategy for multiplex genome engineering, coinciding gene integration which simplified the selection of desired genomic characteristics.
Keywords: Apoptosis, CHO cell, Caspase 7, CRISPR-Associated Protein 9
Copyright and License Information
© 2021 The Authors.
This is an Open Access article distributed under the terms of the Creative Commons Attribution (CC BY), which permits unrestricted use, distribution, and reproduction in any medium, as long as the original authors and source are cited. No permission is required from the authors or the publishers.
Introduction
Producing advantageous genomic characteristics in mammalian cell lines is among the highly precious strategies for gene-functional studies.1,2 Genome editing has been mostly carried out by using traditional methods such as RNA interference3,4 and homologous recombination. Nevertheless, the low frequency of desirable mutants and low specificity in addition to spontaneous cleavage of chromosomal DNA has resulted in the invention of site-specific nucleases. Recently, clusters of regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) systems have opened a promising window for rapid and efficient gene editing at the specific genomic sites.5,6 The CRISPR/Cas9 system is composed of two components: guide RNA (gRNA) and Cas9 protein. Nuclease activity of Cas9 protein induces DNA double-strand breaks (DSBs) in any genomic region which is recognized by a gRNA. This gRNA must be accompanied by an adjacent protospacer motif (PAM) sequence in the target locus.7,8 NGG is a PAM sequence of Streptococcus pyogenes Cas9 (SpCas9) as the most versatile Cas9 protein.9,10
The CRISPR technology has been used for the manipulation of various mammalian cell lines, such as Chinese hamster ovary (CHO) cells. CHO cell line is a primary expression system that is widely utilized for the production of therapeutic pharmaceutics.11 These cells are the host for producing approximately one third of all biopharmaceuticals approved by the Food and Drug Administration (FDA) since 1982.12 Hence, the amelioration of this mammalian expression system is of great commercial interest. In order to respond to the demands of the market for large scale production of biologics, CHO cells must be grown in large bioreactors at high densities. This high density of cells leads to an environmental perturbation, and induces cell stress due to the limitation of nutrients and oxygen as well as the accumulation of toxic metabolic products. Sever and continuous stress prompts cell death by one of the two pathways, including passive cell death called necrosis and apoptosis as programmed cell death.13 Cell death by apoptosis is identified by specific morphological characters and activation of a variety of cellular signaling cascades. All death-inducing pathways need the involvement of specific downstream caspase effectors. Caspases are classified into two groups including initiator caspases (caspases 8, 9, 10 and 12) and executor caspases (caspases 3, 6 and 7). Active caspase 3 and 7 break a large set of substrates, which result in the characteristic morphological and biochemical clues of apoptosis including release of phosphatidylserine, nuclear condensation and genomic DNA fragmentation.14 Findings suggested that caspase-7 is responsible for cell detachment and ROS production.15 Hence, alteration of this gene may interfere with downstream pathways of apoptosis resulting in an apoptosis-resistant cell line. The application of this genome engineered cell line for the expression of therapeutic proteins may enhance the yield and decrease the final cost of biopharmaceutical products.
In the past decade, various engineered CHO cell lines have been established by using CRISPR/Cas9 technology.16 Cas9 nuclease can target multiple-locus due to the coincidental recognition of multiple PAM sequences11. This feature provides an opportunity for multiple recognition of targets by using multiple gRNAs beside a Cas9 protein.17 Since the co-transfection of numerous plasmids can lead to insufficient transfection efficiency of targeted cells, Sakuma and his colleagues developed a smooth and effective construction system called “all-in-one CRISPR/Cas9 systems”. This system, which is composed of vectors, expresses the Cas9 protein as well as up to seven gRNAs that are in tandem ligated into a single vector employing the Golden Gate assembly method.18
Gene silencing mediated by the CRISPR system is achieved via different strategies such as induction of point mutation, CRISPR excision, CRISPR homology-directed repair (CRISPR-HDR), and CRISPR/Cas9 homology-independent targeted integration (CRISPR HITI).
CRISPR excision can be defined as the complete elimination of a DNA segment surrounding an exon. In CRISPR HDR, the desired DNA construct is inserted in the genomic region site specifically by the HDR mechanism. CRISPR HITI employs an NHEJ repair pathway and CRISPR/Cas9 system to replace the genomic sequence located between two DSBs formed by Cas9 with the targeting fragment flanking on the bait vector (Figure 1).19 The invention of HITI facilitates knock out/in genetic modifications of primary and differentiated cells both in vitro and in vivo.20
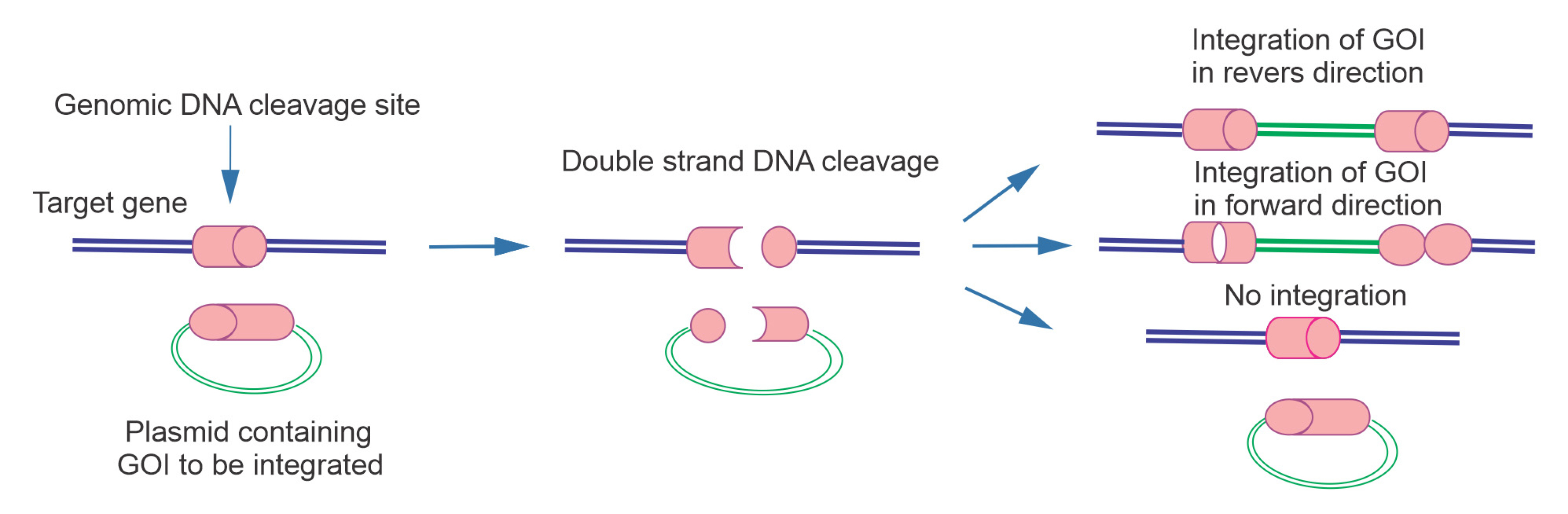
Figure 1.
Scheme of HITI technology, which uses NHEJ-mediated targeted integration. In this method, Cas9 cleaves both genomic sequence and bait vector which harboring gRNA. The gRNA in bait vector is preceded by PAM sequence. This cleavage may repair in 3 conditions include 1: without gene integration 2: with integration in reverse direction (in these conditions DNA undergoes additional cleavage), 3: forward gene integration (gRNA can no longer bind to target cleavage sequence due to errors from NHEJ repair).
.
Scheme of HITI technology, which uses NHEJ-mediated targeted integration. In this method, Cas9 cleaves both genomic sequence and bait vector which harboring gRNA. The gRNA in bait vector is preceded by PAM sequence. This cleavage may repair in 3 conditions include 1: without gene integration 2: with integration in reverse direction (in these conditions DNA undergoes additional cleavage), 3: forward gene integration (gRNA can no longer bind to target cleavage sequence due to errors from NHEJ repair).
In the following lines, we will be describing the protocol for using the combination of the CRISPR multiplex system and CRISPR HITI technology, which resulted in a caspase-7 knockout CHO cell line.
Materials and Methods
Competent E. coli DH5a cells (Pasture Institute, Tehran, Iran) have been used for transformation of plasmids. Adherent CHO-K1 cell line was purchased from Pasture institute, Tehran, Iran.
Chemicals used in this study include Sodium chloride (Merck, USA), Deoxyribonucleic acid sodium salt (Merck, USA), Bacto yeast extract (Merck, USA), Bacto peptone (Thermo Fisher Scientific, USA), Bacto tryptone (Thermo Fisher Scientific, USA), sodium dodecyl sulphate (Merck, USA), hydrochloric acid (Merck, USA), EDTA (Merck, USA), 100% ethanol (Merck, USA), 100% glycerol (Merck, USA).
DNA purification kits (Dena Zist, Mashhad, Iran) and Gene All ExpinTM miniprep kit, (GeneAll, Korea) were the molecular biology reagents that used in this study.
All in one CRISPR system used in this study was containing pX330S-2 (Plasmid #58778, Addgene, USA), pX330A-1x2 (Plasmid #58766, Addgene, USA). We use PX460-1 containing U6 promoter-sgRNA insertion site-sgRNA scaffold and CAG promoter-enhanced GFP (EGFP)-bovine growth hormone polyadenylation signal (Dena Zist Company, Mashhad, Iran) as the HITI vector. The selection markers that use for colony selection were Ampicillin (Sigma-Aldrich, USA) and Spectinomycin hydrochloride (Sigma-Aldrich, USA). Primers used in this study are listed in Table 1.
Table 1.
The sequences of gRNAs and primers
|
Name of primers
|
Sequences
|
| Exon 4 gRNA |
Fwd: CACCgAGATGGCGTGACGCCAATAA
Rew: AAACTTATTGGCGTCACGCCATCTC |
| Exon 4 Bait gRNA |
Fw: caccgCCTTTATTGGCGTCACGCCATCT
Rev: aaacAGATGGCGTGACGCCAATAAAGGc |
| Exon 5 gRNA |
Fwd: CACCGATACGCTTTAGGCATGCCG
Rew: AAACCGGCATGCCTAAAGCGTATC |
| Exon 5 Bait gRNA |
Fw: caccCCTCGGCATGCCTAAAGCGTATC
Rev:aaacGATACGCTTTAGGCATGCCGAGG |
| CRISPR step2 |
Fwd: GCCTTTTGCTGGCCTTTTGCTC
Rew: CGGGCCATTTACCGTAAGTTATGTAACG |
Fast Digest BbsI (BpiI) (Thermo Scientific, USA, FD1014), Fast Digest Eco3I (Bsa1) (Thermo Scientific, USA, FD0294), T4 DNA Ligase (Life Technologies, USA, EL0014), BbsI (BpiI) (Thermo Scientific, USA, EL1012) are the restriction enzymes that have been used for gRNA cloning.
LB medium containing 5 g sodium chloride, 2.5 g Bacto yeast extract, 5 g Bacto tryptone, and up to 500 mL ddH2o was use for liquid bacterial culture. For making solid medium, 1% (w/v) Bacto agar was added and sterilized by heating for 20 minutes at 121°C.
DMEM (Gibco, USA) supplemented with 10% FBS was used for CHO cell culture. For transformation of CHO cells by CRISPR vectors, we applied Fermentas, USA).
gRNA designing for multiplex targeting
The specificity of Cas9 is identified by the gRNA sequence, which works as a guide for Cas9 to cleave the target sequence. In the case of SpCas9, the target sequence must immediately be preceded by a PAM sequence (5′NGG). The 20-nt guide sequence must be complimentary with the opposite strand to trigger Cas9 cleavage at ~3 bp upstream of the PAM. It is worth pointing out that the PAM sequence is needed to be present in the target DNA sequence, but not in the 20-nt guide in the gRNA.21 The specificity of gRNA is a critical issue; hence the selection of gRNAs with minimum off-target activity is necessary.22
Also, the gene targeting strategy is another significant issue. Various strategies have been used for gene targeting by CRISPR systems such as CRISPR HDR, CRISPR excision, and CRISPR HITI. Since the selection of knock out cells becomes a tedious task in CRISPR excision, we employed CRISPR HITI, which does not depend on the existence of homology arms and also makes single-cell selection easy. This technology allows the insertion of transgenes into both proliferating and non-proliferating cells efficiently because it is a cell cycle-independent strategy.20
To this end, the multiplex CRISPR system containing two gRNAs were constructed for targeting two genomic sites on the caspase-7 gene. Furthermore, gRNAs harboring genomic PAM was cloned in bait vectors. Nuclease activity of Cas9 resulted in the DNA cleavages in both genomic sites and bait vectors. Activation of the repair system subsequently integrates the cleaved sequence of bait vector (containing GFP) into a genomic site.
When it comes to the induction of an efficient mutation, the targeting locus is another critical issue. It is recommended to target CDS (coding DNA sequence) regions or the splicing sites of genes or active sites of protein.23,24 In this study, to augment the precision of gene disruption, the active site of caspase-7 flanking between exon 4 and exon 5 was targeted.
In order to select an appropriate gRNA with the minimum off-target activity, various on line soft wares with diverse sensitivity were used (Table 2). Desired loci were used as an input for these soft wares. Outputs with the highest on-target activity and the lowest unintended effects were selected. Because the U6 RNA polymerase III promoters (flanked in PX330 vector) prefers a guanine (G) nucleotide as the first base of its transcript, so in order to express the gRNA an extra G is added at the 5′ end where the guide sequence does not begin with G.25
Table 2.
gRNA designing soft wares
Plasmid construction and cloning of gRNAs
Annealing the oligonucleotides
To generate the CRISPR/Cas9 plasmids against caspase-7, top and bottom oligonucleotides were synthesized in a final concentration of 100 μM and phosphorylated and annealed in the mixture containing gRNA top and bottom (1 μL), T4 ligation buffer (1 μL), T4 PNK 1 μL and ddH2O (6 μL).
The reaction tube containing the mixture was put in the thermocycler with the following characteristics: 37°C (30 minutes); 95°C (5 minutes); ramp down to 25°C at 5°C min–1. The dilution of phosphorylated and annealed gRNAs was carried out by adding 1 μL of oligo to 199 μL of deionized water.
Cloning of gRNAs in all in one multiplex vector
The annealed oligonucleotides, pX330A 1-2/S-2 vectors, BpiI enzyme, Quick ligase, and T4 DNA ligase buffer were mixed in two steps including digestion and ligation for cloning of gRNAs in the target vectors. The digestion reaction mixture was containing pX330A 1-2/S-2 (8.5 μL), BpiI (0.5 μL) and 10x buffer (1 μL). This mixture was prepared and maintained at 37°C for 10 minutes. The ligation reaction was performed by using digested vector (10 μL), 10 µM annealed gRNA (5 μL), T4 DNA ligase buf (2 μL), Quick ligase (1 μL) and ddH2O (2 μL).
The mentioned mixture was transferred to a thermal cycler with reactions including 3 cycles of 37°C (5 minutes) and 16°C (10 minutes). Following the cycling reaction, extra BpiI digestion was carried out by adding BpiI (0.5 μL) and ddH2O (9.5 μL) to the previous tube and maintaining at 37°C for 1 hour. A list of the constructed plasmids with their contents is represented in Table 3.
Table 3.
constructed vectors containing related gRNAs
|
Vectors
|
Targeted locus
|
gRNA sequence
|
| pX330 S-2 |
Exon 4 |
Fwd: CACCgAGATGGCGTGACGCCAATAA |
| Rew: AAACTTATTGGCGTCACGCCATCTC |
| pX330A1-2 |
Exon 5 |
Fwd: CACCGATACGCTTTAGGCATGCCG |
| Rew: AAACCGGCATGCCTAAAGCGTATC |
Golden Gate cloning
Golden Gate cloning was carried out by using pX330S-2 and pX330A1x2 plasmids 3μL, BsaI-HF enzyme (1 μL), Quick ligase (1 μL), T4 DNA ligase buffer (2 μL) and ddH2O (11 μL) in a single tube. This mixture was harbored a thermal cycling reaction including 4 cycle 37°C (15 minutes) and 16°C (30 minutes). Subsequent to the cycling reaction, extra BsaI-HF digestion was carried out at 37°C for 10 minutes (Figure 2).
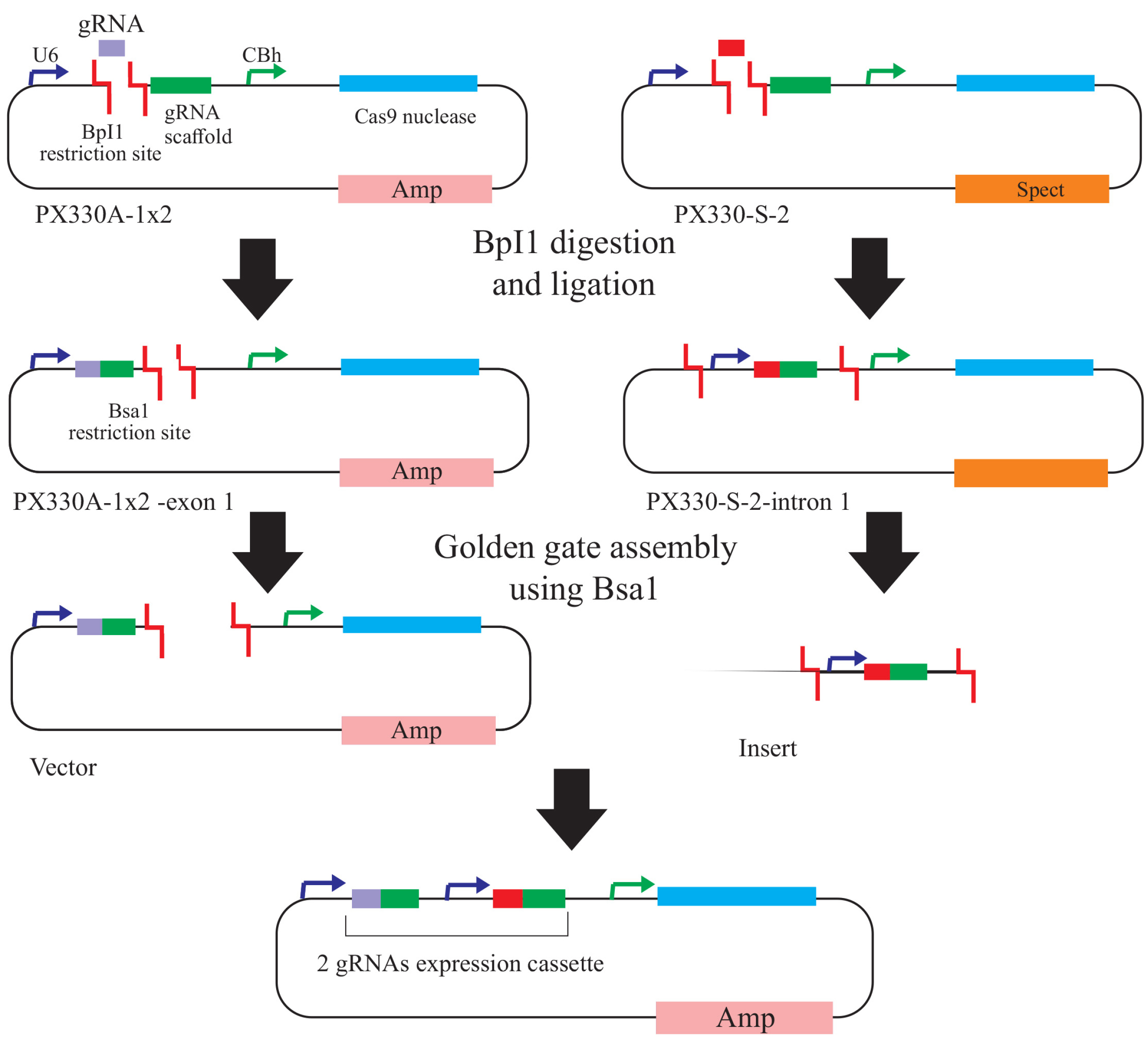
Figure 2.
Schematic overview of the all-in-one CRISPR/Cas9 vector construction system for multiplex targeting of genome. STEP 1: Oligonucleotides relating to each target sequence are annealed and inserted into pX330A1-2 or pX330S-2 vectors by BpiI digestion. STEP 2: The constructed vectors bearing single gRNA expression cassettes are then assembled into an all-in-one vector containing multiple gRNA cassettes using the Golden Gate assembly method. Abbreviations: Amp, ampicillin; Spec, spectinomycin; U6, human U6 promoter; CBh, chicken beta-actin short promoter.
.
Schematic overview of the all-in-one CRISPR/Cas9 vector construction system for multiplex targeting of genome. STEP 1: Oligonucleotides relating to each target sequence are annealed and inserted into pX330A1-2 or pX330S-2 vectors by BpiI digestion. STEP 2: The constructed vectors bearing single gRNA expression cassettes are then assembled into an all-in-one vector containing multiple gRNA cassettes using the Golden Gate assembly method. Abbreviations: Amp, ampicillin; Spec, spectinomycin; U6, human U6 promoter; CBh, chicken beta-actin short promoter.
The confirmation of plasmid construction was carried out by colony polymerase chain reaction (PCR) utilizing step2-F and step2-R primers (Table 1), followed by agarose gel electrophoresis.
Cloning of bait gRNAs in CRISPR HITI vectors
The PX460-1 vector (2 μL), BpiI enzyme (1 μL), Gbuffer (1 μL) and ddH2O (16 μL) were mixed and maintained in 37°C over night to digest the vector and then used for ligation as follow. The ligation mixture was contained Digested pX460-1 vector (0.5 μL), 10 µM annealed gRNA (0.5 μL), 10×T4 DNA ligase buf (1 μL), Ligase buf (2 μL), ddH2O (16 μL). The mentioned mixture was maintained at 22°C overnight to ligate the bait gRNAs with the pX460-1 vector.
After each cloning, resulted plasmids were transformed into the competent DH5a strain, which followed by colony PCR to identified desired plasmids.
Cell culture and transfections
CHOK1 cells were grown in DMEM medium supplemented with 10% FBS and antibiotics. Cells were incubated in a humidified incubator at 37°C and 5% CO2. One day before the transfection, cells were seeded in 6 well plates at 5-6 × 105 density in antibiotic-free media. Cells were transfected with all in one vector encoding Cas9 and 2 gRNAs targeting the exons 4 and 5 of caspase-7 and bait vectors containing bait gRNAs and EGFP expression cassette. Each well was transfected with 3 μg DNA, respectively, using Lipofectamine 2000 with OptiMEM medium as transfection reagent based on the manufacturer’s recommendations. PmaxGFP® vector (Lonza, Basel, Switzerland) was used as control transfection reagent for confirmation of transfection efficiencies.
Selection and detection of knockout alleles
A week after transfection, cell sorting was carried out by using FACS BD instrument. GFP expressing cells were seeded in 96 well plate by serial dilution to achieve a single cell clone. Following several passages, genomic DNA isolation and analysis was carried out for both knockout and wild-type cell lines followed by PCR amplification. Forward strand of exon 4 gRNA was used as a forward primer, and step2 R primer and the reverse strand of exon 5 gRNA were used as a reverse primers. PCR- products after clean-up and recovery reactions, were subjected to Sanger sequencing.
Results
Plasmid construction and cloning of sgRNA
Agarose gel electrophoresis followed by sequencing was applied to confirm the cloning of gRNAs within all in one and bait vectors. The cloning of exon 5 gRNA in Px330-A1-2 resulted in a PCR product with a length of 290 and 680 bp (Figure 3a). The cloning of exon 4 gRNA in PX330-s2 produced products with a length of 221 and 311 bp (Figure 3b). The construction of all in one vector confirmed by observing the PCR products with a length of 458bp and 226bp (Figure 3c). The PCR product validated the cloning of bait gRNAs in HITI vectors with a length of 301 bp (Figure 3d).
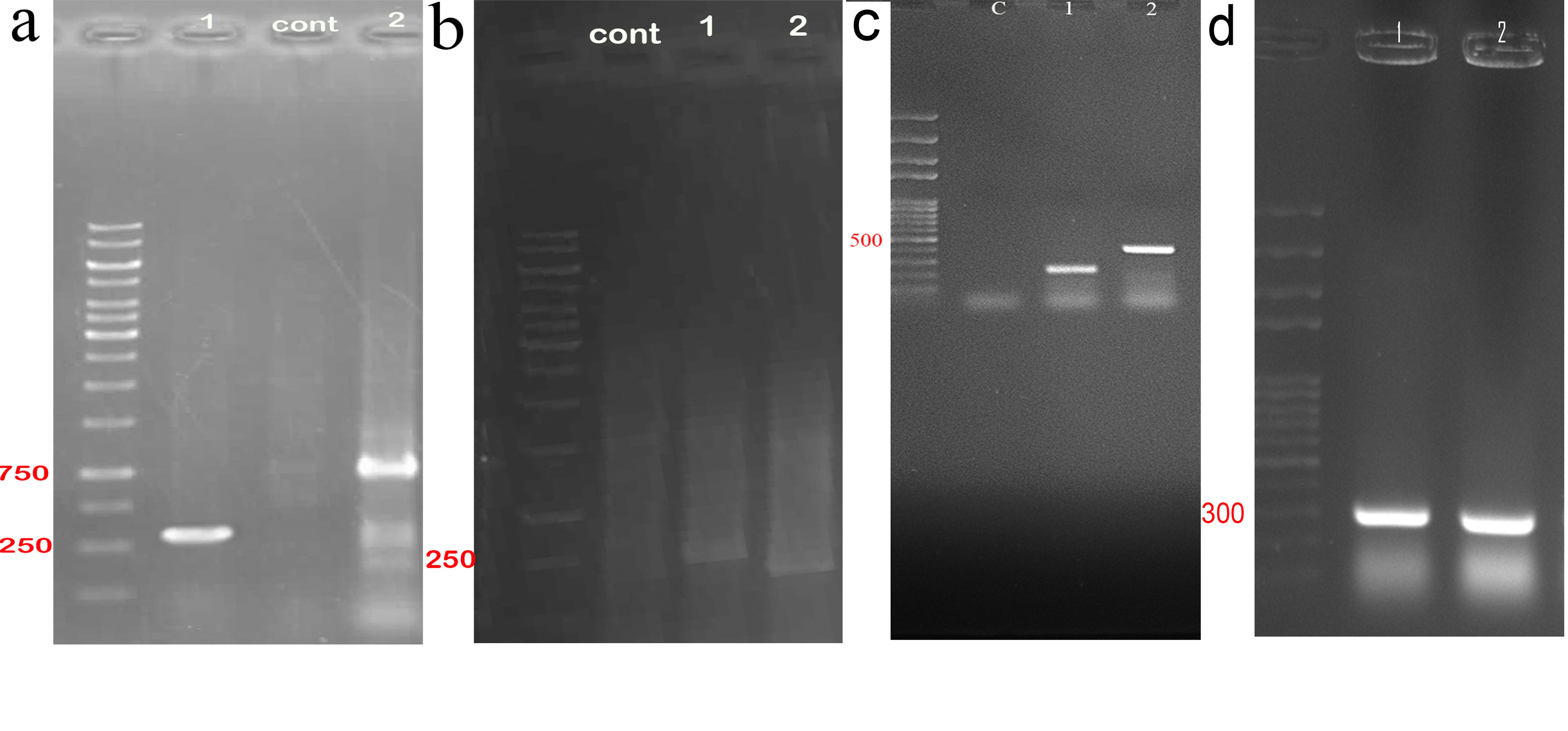
Figure 3.
Gel electrophoresis of PCR product related to sgRNAs cloning in all in one and bait vectors. (a) exon 5 gRNA was cloned in PX330A 1x2, PCR products with the length of 229bp (lane 1 by using: step2 forward primer and reverse strand of exon 5 gRNA) and 680 bp (lane 3 by using: forward strand of exon 5 gRNA and step 2 reverse primer) confirmed this insertion. (b) exon 4 gRNA was cloned in PX330-s2, this insertion was validated by PCR product with the length of 229 bp (lane 1 by using: step2 forward primer and reverse strand of exon 5 gRNA) and 311bp (lane 2: by using forward strand of exon 4 gRNA and step 2 reverse primer). (c) construction of all-in-one vector was confirmed with the observation of 229 bp (lane 1 by using: step2 forward primer and reverse strand of exon 5 gRNA) and 458 bp (lane 1 by using: step2 forward primer and reverse strand of exon 4 gRNA) bands showed the presentation of both gRNAs in this vector. (d) this image refers to construction of bait vectors which contains each bait gRNA with the PCR product of 301 bp (lane 1 ,2 by using each forward strand of bait gRNAs and step 2 reverse primer).
.
Gel electrophoresis of PCR product related to sgRNAs cloning in all in one and bait vectors. (a) exon 5 gRNA was cloned in PX330A 1x2, PCR products with the length of 229bp (lane 1 by using: step2 forward primer and reverse strand of exon 5 gRNA) and 680 bp (lane 3 by using: forward strand of exon 5 gRNA and step 2 reverse primer) confirmed this insertion. (b) exon 4 gRNA was cloned in PX330-s2, this insertion was validated by PCR product with the length of 229 bp (lane 1 by using: step2 forward primer and reverse strand of exon 5 gRNA) and 311bp (lane 2: by using forward strand of exon 4 gRNA and step 2 reverse primer). (c) construction of all-in-one vector was confirmed with the observation of 229 bp (lane 1 by using: step2 forward primer and reverse strand of exon 5 gRNA) and 458 bp (lane 1 by using: step2 forward primer and reverse strand of exon 4 gRNA) bands showed the presentation of both gRNAs in this vector. (d) this image refers to construction of bait vectors which contains each bait gRNA with the PCR product of 301 bp (lane 1 ,2 by using each forward strand of bait gRNAs and step 2 reverse primer).
CHO-K1 transfection, clonal selection and knockout clone detection
Transfection efficiency of CRISPR plasmids was ascertained by CytationTM 5 Cell Imaging. The results of this imaging demonstrated that 70% of cells had been transfected successfully (Figure 4).
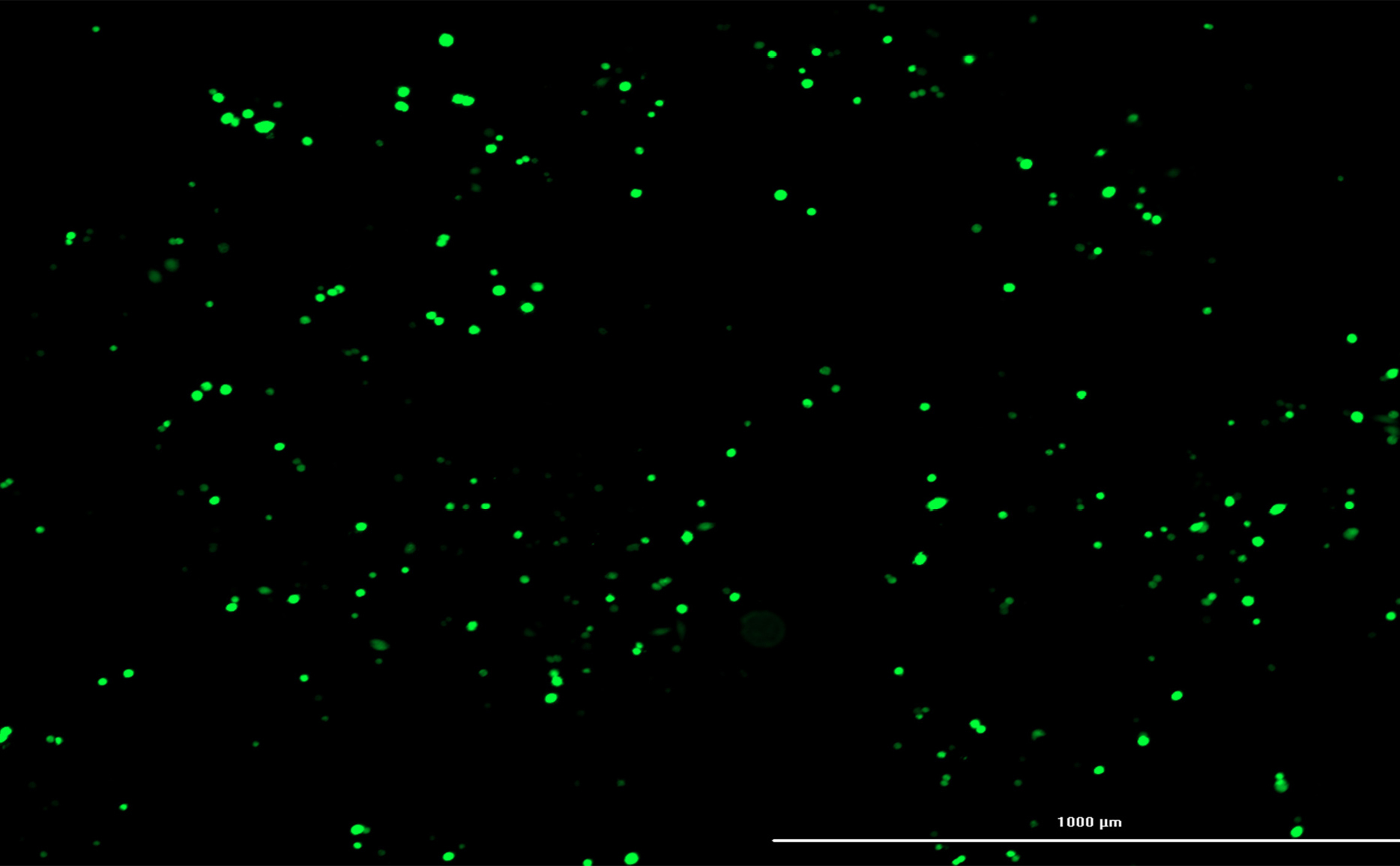
Figure 4.
Transfection efficiency. CytationTM 5 Cell Imaging showed approximately 70% transfection efficiency.
.
Transfection efficiency. CytationTM 5 Cell Imaging showed approximately 70% transfection efficiency.
Cell sorting (by using FACSCALIBUR, BD, USA) followed by single-cell cloning via serial dilution was used for clonal selection. Detection of knockout cells which expressed GFP was carried out by genomic DNA isolation and PCR for each single clone. EGFP integration was confirmed by observing a 220 bp band on gel electrophoresis, which belonged to the bait vector. Following the first round of transfection, results of PCR and gel electrophoresis showed that the selected clones were heterozygote by displaying both 395 bp band (which belonged to native genomic allele) and 220 bp (which belonged to knockout allele) PCR product (Figure 5a). From these clones, EGFP expressing clone (Figure 6) was selected and harbored the second round of transfection. The results of genomic PCR following the second transfection were similar to the first transfection and resulted in heterozygote caspase-7 knockout clone (Figure 5b). The findings of PCR product sequencing also confirmed the heterozygosity of clones (Figure 7a and 7b).
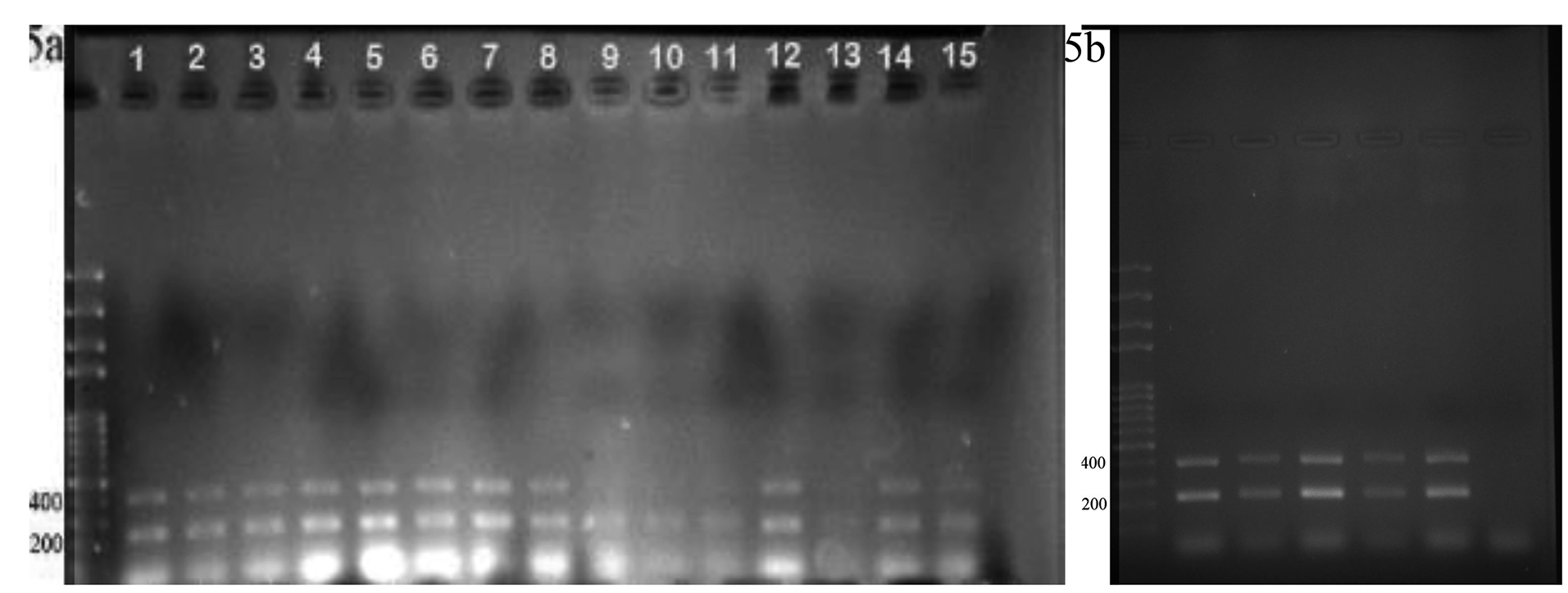
Figure 5.
The confirmation of heterozygote knocked out allele. (a) This gel electrophoresis showed
heterozygote clones contain one allele of wild type cells with the length of 395 bp and knock out
allele with 220 bp length. (b) the results of second round of transfection represent the both 395 and 220 bp PCR product which confirmed the results of first round transfection.
.
The confirmation of heterozygote knocked out allele. (a) This gel electrophoresis showed
heterozygote clones contain one allele of wild type cells with the length of 395 bp and knock out
allele with 220 bp length. (b) the results of second round of transfection represent the both 395 and 220 bp PCR product which confirmed the results of first round transfection.
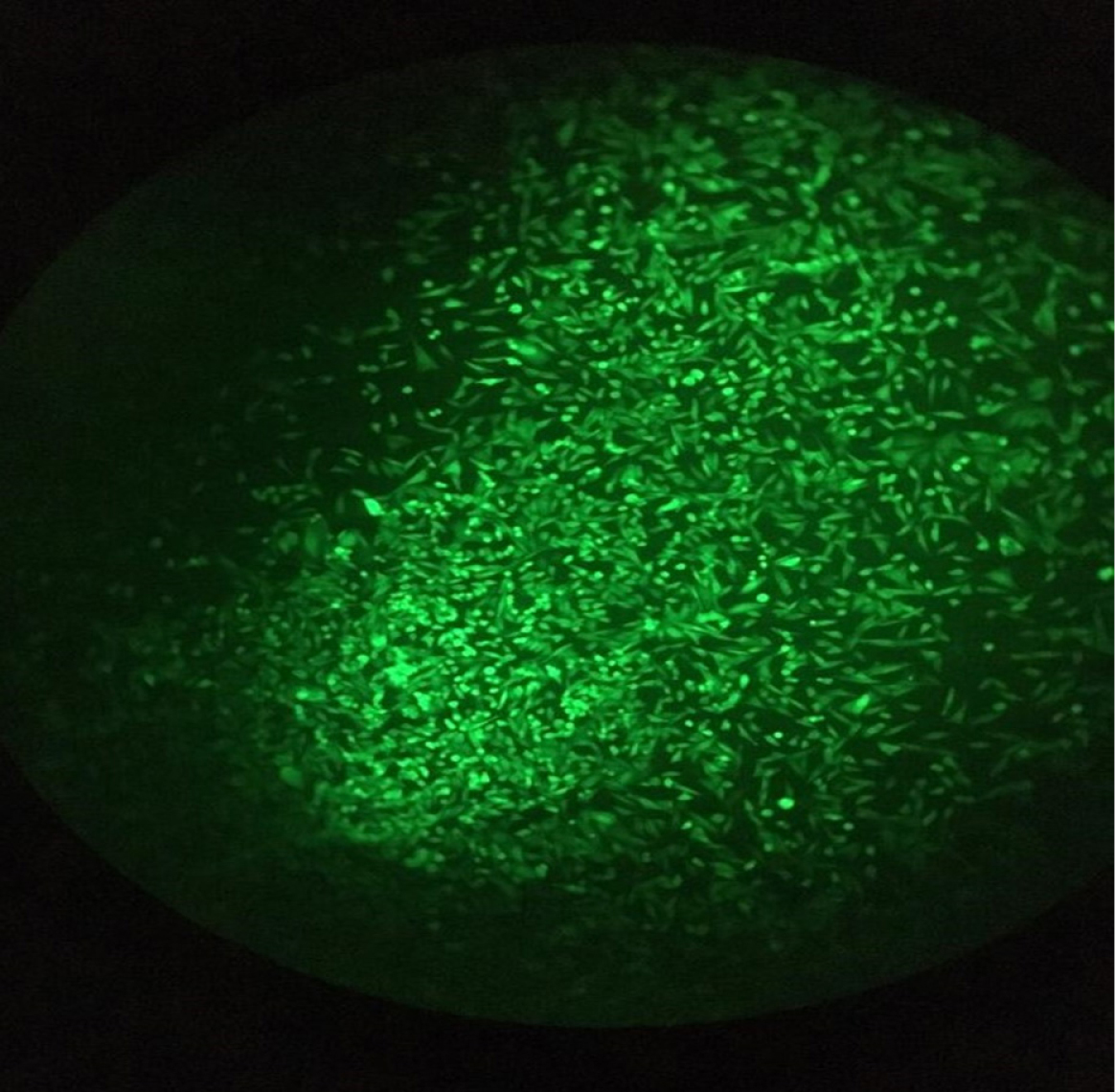
Figure 6.
Heterozygote caspase 7 knock out CHO cell line expressing EGFP.
.
Heterozygote caspase 7 knock out CHO cell line expressing EGFP.
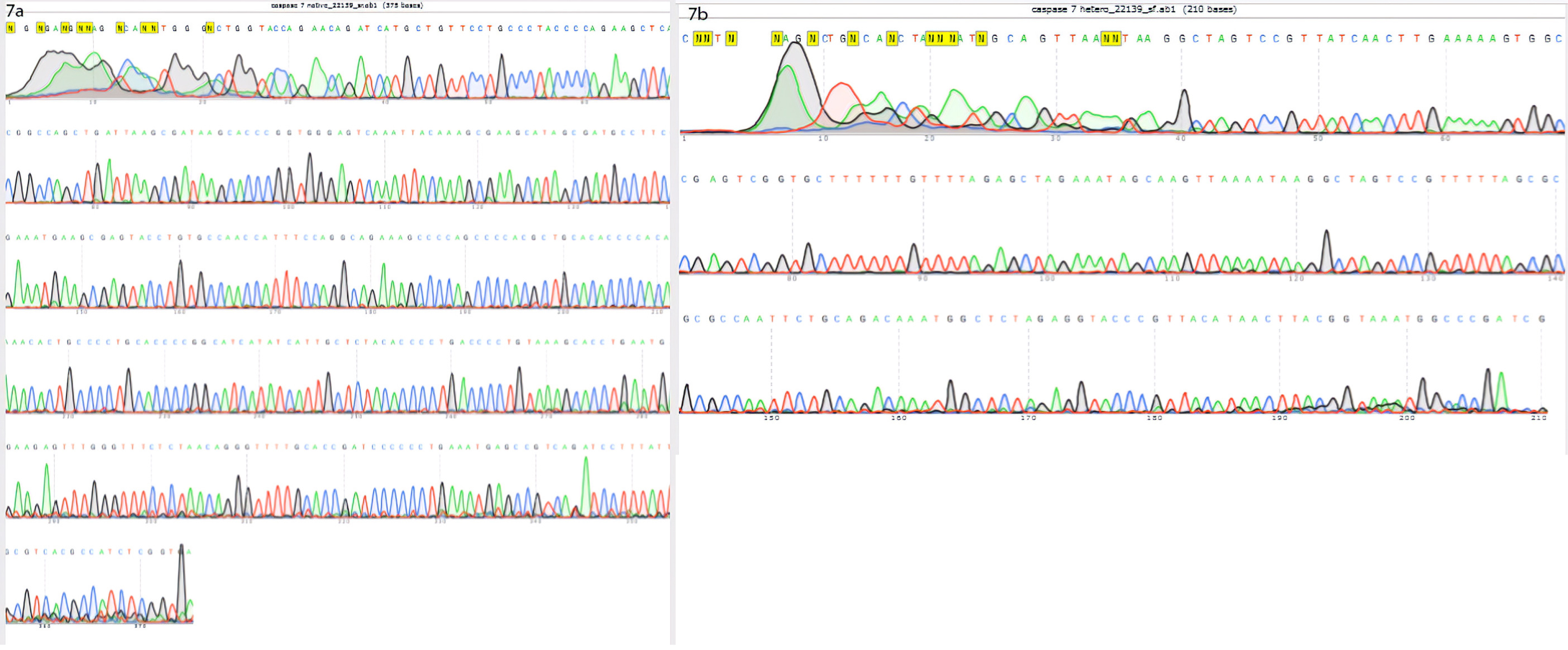
Figure 7.
Findings of sequencing are represented in these figures: (a) the results of 395 bp PCR product sequencing showed the intact allele of CHO cell. (b) the results of 220 bp PCR product sequencing showed the integration of bait vector in one allele of CHO cell genome
.
Findings of sequencing are represented in these figures: (a) the results of 395 bp PCR product sequencing showed the intact allele of CHO cell. (b) the results of 220 bp PCR product sequencing showed the integration of bait vector in one allele of CHO cell genome
Discussion
Genome editing using the CRISPR/Cas9 system is an efficient strategy for the establishing of genome-engineered cells. Multiplex genome editing is one of the most appealing approaches in gene modification today. This approach provides us with a simple and effective strategy for single vector that mediated multiple targeting.26 In order to produce knockout cells, various strategies have been at our disposal, including the CRISPR excision, CRISPR HITI and CRISPR HDR. For the execution of CRISPR excision, no donor vectors were used, and therefore no reporter genes were observed to be integrated into the genome. Hence, the selection of knockout cells becomes a repetitive task. In contrast, CRISPR HDR is executed by recruiting donor vectors thus allow the selection of the targeted cells based on resistance to antibiotic or observing fluorescence. It is noteworthy that this strategy requires the homology arms, which must be cloned into the bait vector. The CRISPR HITI does not depend on the existence of homology arms and facilitates the selection of the knockout cells. HITI approach empowers us to the direct invention of knockout cells; thus each cell with fluorescence phenotype is easily selected as an allele knockout.19,27
In comparison with the HDR, HITI is more eventful as it relies on the cell cycle phase independent mechanism of double-stranded DNA break repair (NHEJ). HITI provides a large percentage of cells with the desired DNA insertion in the correct orientation and a correct locus. However, certain limitations are recognized in HITI. Due to the design procedure, inevitably, additional nucleotides are introduced into the target locus accompanied by the insertion of the DNA template at the ends of the insert. Hence, special consideration should be paid to possible reading frame shifts as well as deletion of native or emergence of new regulatory sequences in the noncoding genomic locus.28
It is widely believed that caspase-7 is an executioner caspase with a role in the earlier stages of the apoptotic pathway. Findings have shown that the deletion of caspase-7 in different cell lines resulted in different cellular micro-environments. For example, disruption of caspase-7 is not lethal in chicken B cell lymphoma line DT40 and mouse embryonic fibroblasts cells.29,30 Nevertheless, the caspase-7 gene deletion is reportedly lethal in mice.31,32 On the same token, we found that homozygote caspase-7 deletion seemed to be lethal in the CHO cell line, which begets more study. These results raise the possibility that the effect of caspase-7 deficiency varies and cell line dependent.
Conclusion
In conclusion, we delineated here the CRISPR/Cas9 mediated strategy that provides caspase-7 knockout CHO cell line. All in one CRISPR/Cas9 vectors facilitate the one-step generation of the mutant organism with multiple engineered genes. Furthermore, the HITI strategy is easy to use and can be employed for the production of knockout cells for a specific gene of interest. CHO cells are the popular mammalian workhorse for the generation of commercial therapeutically essential proteins. The development of recombinant CHO has become a priority in biopharmaceutical manufacture. Although, the resulted CHO caspase-7 knockout cell line in this study was a heterozygote, efforts are ongoing to attain a probable homozygote knockout cell. This can be materialized by using another selection marker such as puromycin, which is integrated into the target site by HITI strategy.
Ethical Issues
Ethics approval for the study was obtained from an ethics committee of Tabriz University of Medical Sciences dated 2016/13/08, No: 5/4/46151.
Conflict of Interest
The authors declare that they have no conflict of interest.
Acknowledgments
Authors would like to thank the Department of Medical Biotechnology, Faculty of Advanced Medical Sciences, Tabriz University of Medical Sciences for supporting this project (grant No. 94/4-2/4). In addition, this study was financially supported by grant No: 950609 of the Biotechnology Development Council of the Islamic Republic of Iran.
References
- Safari F, Rahmani Barouji S, Tamaddon AM. Strategies for improving siRNA-induced gene silencing efficiency. Adv Pharm Bull 2017; 7(4):603-9. doi: 10.15171/apb.2017.072 [Crossref] [ Google Scholar]
- Safari F, Tamaddon AM, Zarghami N, Abolmali S, Akbarzadeh A. Polyelectrolyte complexes of hTERT siRNA and polyethyleneimine: effect of degree of PEG grafting on biological and cellular activity. Artif Cells Nanomed Biotechnol 2016; 44(6):1561-8. doi: 10.3109/21691401.2015.1064936 [Crossref] [ Google Scholar]
- Zarredar H, Ansarin K, Baradaran B, Shekari N, Eyvazi S, Safari F. Critical microRNAs in lung cancer: recent advances and potential applications. Anticancer Agents Med Chem 2018; 18(14):1991-2005. doi: 10.2174/1871520618666180808125459 [Crossref] [ Google Scholar]
- Zarredar H, Pashapour S, Farajnia S, Ansarin K, Baradaran B, Ahmadzadeh V. Targeting the KRAS, p38α, and NF-κB in lung adenocarcinoma cancer cells: the effect of combining RNA interferences with a chemical inhibitor. J Cell Biochem 2019; 120(6):10670-7. doi: 10.1002/jcb.28357 [Crossref] [ Google Scholar]
- Eisenhut P, Klanert G, Weinguny M, Baier L, Jadhav V, Ivansson D. A CRISPR/Cas9 based engineering strategy for overexpression of multiple genes in Chinese hamster ovary cells. Metab Eng 2018; 48:72-81. doi: 10.1016/j.ymben.2018.05.017 [Crossref] [ Google Scholar]
- Safari F, Sharifi M, Farajnia S, Akbari B, Karimi Baba Ahmadi M, Negahdaripour M. The interaction of phages and bacteria: the co-evolutionary arms race. Crit Rev Biotechnol 2020; 40(2):119-37. doi: 10.1080/07388551.2019.1674774 [Crossref] [ Google Scholar]
- Safari F, Farajnia S, Arya M, Zarredar H, Nasrolahi A. CRISPR and personalized Treg therapy: new insights into the treatment of rheumatoid arthritis. Immunopharmacol Immunotoxicol 2018; 40(3):201-11. doi: 10.1080/08923973.2018.1437625 [Crossref] [ Google Scholar]
- Safari F, Hatam G, Behzad-Behbahani A, Rezaei V, Barekati-Mowahed M, Petramfar P. CRISPR system: a high-throughput toolbox for research and treatment of Parkinson’s disease. Cell Mol Neurobiol 2020; 40(4):477-93. doi: 10.1007/s10571-019-00761-w [Crossref] [ Google Scholar]
- Safari F, Farajnia S, Ghasemi Y, Zarghami N. New developments in CRISPR technology: improvements in specificity and efficiency. Curr Pharm Biotechnol 2017; 18(13):1038-54. doi: 10.2174/1389201019666180209120533 [Crossref] [ Google Scholar]
- Safari F, Zare K, Negahdaripour M, Barekati-Mowahed M, Ghasemi Y. CRISPR Cpf1 proteins: structure, function and implications for genome editing. Cell Biosci 2019; 9:36. doi: 10.1186/s13578-019-0298-7 [Crossref] [ Google Scholar]
- Grav LM, Lee JS, Gerling S, Kallehauge TB, Hansen AH, Kol S. One-step generation of triple knockout CHO cell lines using CRISPR/Cas9 and fluorescent enrichment. Biotechnol J 2015; 10(9):1446-56. doi: 10.1002/biot.201500027 [Crossref] [ Google Scholar]
- Walsh G. Biopharmaceutical benchmarks 2014. Nat Biotechnol 2014; 32(10):992-1000. doi: 10.1038/nbt.3040 [Crossref] [ Google Scholar]
- Hwang SO, Lee GM. Nutrient deprivation induces autophagy as well as apoptosis in Chinese hamster ovary cell culture. Biotechnol Bioeng 2008; 99(3):678-85. doi: 10.1002/bit.21589 [Crossref] [ Google Scholar]
- Lamkanfi M, Kanneganti TD. Caspase-7: a protease involved in apoptosis and inflammation. Int J Biochem Cell Biol 2010; 42(1):21-4. doi: 10.1016/j.biocel.2009.09.013 [Crossref] [ Google Scholar]
- Krampe B, Al-Rubeai M. Cell death in mammalian cell culture: molecular mechanisms and cell line engineering strategies. Cytotechnology 2010; 62(3):175-88. doi: 10.1007/s10616-010-9274-0 [Crossref] [ Google Scholar]
- Xiong K, Marquart KF, la Cour Karottki KJ, Li S, Shamie I, Lee JS. Reduced apoptosis in Chinese hamster ovary cells via optimized CRISPR interference. Biotechnol Bioeng 2019; 116(7):1813-9. doi: 10.1002/bit.26969 [Crossref] [ Google Scholar]
- Sakuma T, Nishikawa A, Kume S, Chayama K, Yamamoto T. Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Sci Rep 2014; 4:5400. doi: 10.1038/srep05400 [Crossref] [ Google Scholar]
- Sakuma T, Sakamoto T, Yamamoto T. All-in-one CRISPR-Cas9/FokI-dCas9 vector-mediated multiplex genome engineering in cultured cells. Methods Mol Biol 2017; 1498:41-56. doi: 10.1007/978-1-4939-6472-7_4 [Crossref] [ Google Scholar]
- Suzuki K, Tsunekawa Y, Hernandez-Benitez R, Wu J, Zhu J, Kim EJ. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016; 540(7631):144-9. doi: 10.1038/nature20565 [Crossref] [ Google Scholar]
- Zare K, Shademan M, Ghahramani Seno MM, Dehghani H. CRISPR/Cas9 knockout strategies to ablate CCAT1 lncRNA gene in cancer cells. Biol Proced Online 2018; 20:21. doi: 10.1186/s12575-018-0086-5 [Crossref] [ Google Scholar]
- Liang X, Potter J, Kumar S, Ravinder N, Chesnut JD. Enhanced CRISPR/Cas9-mediated precise genome editing by improved design and delivery of gRNA, Cas9 nuclease, and donor DNA. J Biotechnol 2017; 241:136-46. doi: 10.1016/j.jbiotec.2016.11.011 [Crossref] [ Google Scholar]
- Bolukbasi MF, Gupta A, Wolfe SA. Creating and evaluating accurate CRISPR-Cas9 scalpels for genomic surgery. Nat Methods 2016; 13(1):41-50. doi: 10.1038/nmeth.3684 [Crossref] [ Google Scholar]
- Gagnon JA, Valen E, Thyme SB, Huang P, Akhmetova L, Pauli A. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS One 2014; 9(5):e98186. doi: 10.1371/journal.pone.0098186 [Crossref] [ Google Scholar]
- Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M, Smith I. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol 2014; 32(12):1262-7. doi: 10.1038/nbt.3026 [Crossref] [ Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013; 8(11):2281-308. doi: 10.1038/nprot.2013.143 [Crossref] [ Google Scholar]
- Kabadi AM, Ousterout DG, Hilton IB, Gersbach CA. Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Res 2014; 42(19):e147. doi: 10.1093/nar/gku749 [Crossref] [ Google Scholar]
- Suzuki K, Izpisua Belmonte JC. In vivo genome editing via the HITI method as a tool for gene therapy. J Hum Genet 2018; 63(2):157-64. doi: 10.1038/s10038-017-0352-4 [Crossref] [ Google Scholar]
- Karagyaur MN, Rubtsov YP, Vasiliev PA, Tkachuk VA. Practical recommendations for improving efficiency and accuracy of the CRISPR/Cas9 genome editing system. Biochemistry (Mosc) 2018; 83(6):629-42. doi: 10.1134/s0006297918060020 [Crossref] [ Google Scholar]
- Korfali N, Ruchaud S, Loegering D, Bernard D, Dingwall C, Kaufmann SH. Caspase-7 gene disruption reveals an involvement of the enzyme during the early stages of apoptosis. J Biol Chem 2004; 279(2):1030-9. doi: 10.1074/jbc.M306277200 [Crossref] [ Google Scholar]
- Masud A, Mohapatra A, Lakhani SA, Ferrandino A, Hakem R, Flavell RA. Endoplasmic reticulum stress-induced death of mouse embryonic fibroblasts requires the intrinsic pathway of apoptosis. J Biol Chem 2007; 282(19):14132-9. doi: 10.1074/jbc.M700077200 [Crossref] [ Google Scholar]
- Zheng TS, Hunot S, Kuida K, Flavell RA. Caspase knockouts: matters of life and death. Cell Death Differ 1999; 6(11):1043-53. doi: 10.1038/sj.cdd.4400593 [Crossref] [ Google Scholar]
- Zheng TS, Hunot S, Kuida K, Momoi T, Srinivasan A, Nicholson DW. Deficiency in caspase-9 or caspase-3 induces compensatory caspase activation. Nat Med 2000; 6(11):1241-7. doi: 10.1038/81343 [Crossref] [ Google Scholar]