Advanced pharmaceutical bulletin. 15(3):678-685.
doi: 10.34172/apb.025.43440
Original Article
Continuous Uptake of MiR-181a-2 Mimetic Induces Constitutive Overexpression of Cellular MiR-181a-2 in MCF-7 Breast Cancer Cells: Links with Progression of Drug Resistance
Olga Evgen’evna Andreeva Formal analysis, Investigation, Visualization, Writing – review & editing, 1, # 
Danila Vladimirovich Sorokin Investigation, 1, #
Svetlana Vladimirovna Vinokurova Data curation, Formal analysis, Investigation, Methodology, Resources, Software, Validation, Visualization, 1
Pavel Borisovich Kopnin Data curation, Formal analysis, Investigation, Methodology, Resources, Validation, 1
Nadezhda Viacheslavovna Elkina Investigation, 1
Danila Sergeevich Elkin Investigation, 1
Maria Dmitrievna Fedorova Investigation, 1
Alexander Mikhailovich Scherbakov Data curation, Formal analysis, Investigation, Resources, Software, Validation, Visualization, Writing – review & editing, 1, 2, *
Mikhail Aleksandrovich Krasil’nikov Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Writing – original draft, 1, *
Author information:
1N.N. Blokhin National Medical Research Center of Oncology, the Ministry of Health of Russia, Moscow, Russia
2Gause Institute of New Antibiotics, Moscow, Russia
#Both authors contributed equally to this work.
Abstract
Purpose:
The aim of this study was to elucidate the mechanisms underlying the formation and maintenance of drug resistance in cancer cells. Previously, we demonstrated that prolonged treatment of estrogen-dependent MCF-7 breast cancer cells with exosomes derived from estrogen-resistant MCF-7/T cells leads to a partial loss of estrogen sensitivity in MCF-7 cells. Moreover, repeated transfection with one of the exosomal microRNAs—microRNA-181a-2—induced an irreversible decrease in hormonal sensitivity in the recipient cells. In the present work, to further investigate the possible mechanism of miR-181a-2-induced acquired resistance, we analyzed the effect of multiple miR-181a-2 transfections on the expression of cellular miR-181a-2 and related signaling proteins.
Methods:
miR-181a-2 was ectopically expressed by mimetic transfection or suppressed by antisense oligonucleotides. miR-181a-2 precursor/MIR181A2HG expression (qRT-PCR) and MIR181A2 locus copy number (qPCR) were assessed. wtSnail was expressed via transient transfection. Tamoxifen sensitivity was measured by MTT assay. Protein expression was studied by immunoblotting, estrogen receptor α/Snail transcriptional activity was evaluated by reporter analysis.
Results:
We found that multiple transfections with miR-181a-2 resulted in a marked increase in cellular miR-181a-2 precursor levels, whereas single transfection had no such effect. Similarly, stable transfection with miR-181a-2 led to increased levels of cellular miR-181a-2 and its host gene, MIR181A2HG, which was associated with partial resistance to tamoxifen. Analysis of the genomic DNA encoding miR-181a-2 revealed no changes in copy number in transfected cells. Furthermore, we identified the transcription factor Snail as a key mediator of miR-181a-2–induced resistance and demonstrated its role in the formation of an autoregulatory loop of miR181a-2 and the maintenance of cell resistance.
Conclusion:
Overall, these results reveal a novel mechanism of resistance-associated signaling pathway rearrangement based on the formation of a miR-181a-2 autoregulatory loop.
Keywords: Breast cancer, MCF-7, MicroRNA, MiR-181a-2, Snail, Tamoxifen resistance
Copyright and License Information
© 2025 The Author (s).
This is an Open Access article distributed under the terms of the Creative Commons Attribution (CC BY), which permits unrestricted use, distribution, and reproduction in any medium, as long as the original authors and source are cited. No permission is required from the authors or the publishers.
Introduction
Despite the proven effectiveness of hormone therapy, its clinical application is limited by the development of tumor resistance to hormones, which can be either primary or acquired, i.e., emerging during hormonal treatment.1 The mechanisms underlying hormonal resistance are well studied: resistance can arise due to an irreversible blockade of hormonal signaling (typically via suppression of the activity or expression of specific intracellular hormone receptors) or via activation of growth-regulating signaling pathways that bypass hormone-dependent signaling.2-5
In addition to reduced receptor levels, major factors contributing to hormonal resistance include: (i) an imbalance between receptor coactivators and corepressors; (ii) ligand-independent receptor activation; and (iii) stimulation of hormone-independent growth pathways (primarily mediated by tyrosine kinase receptors) that sustain tumor growth in the absence of hormones.
Primary resistance is typically associated with mutations in specific genes that disrupt hormonal signaling (e.g., repression of hormone receptors) and/or activation of hormone-independent growth signals (e.g., tyrosine kinase cascades). In contrast, acquired resistance is mainly driven by epigenetic mechanisms. Among these, microRNAs (miRNAs) play a particularly important role, acting directly as epigenetic regulators of gene expression and indirectly via modulation of DNA methylation and histone acetylation.6
To date, the involvement of miRNAs in hormonal resistance of tumors has been well documented. These include miRNAs negatively regulating estrogen receptor α (ERα) or its coactivator/corepressor proteins, as well as miRNAs controlling signaling proteins in the ERα cascade (HER2, EGFR, Akt, MAPK) and tumor suppressors.7-16
In recent years, increasing attention has been paid to resistance mediated by exosomes—small extracellular vesicles secreted by cells that can be incorporated into recipient cells. In our earlier studies on estrogen-dependent MCF-7 breast cancer cells and the tamoxifen-resistant MCF-7/T subline, we demonstrated that exosomes derived from resistant cells can transfer hormonal resistance to parental cells.17,18
Here, we show that stable overexpression of one exosomal miRNA—miR-181a-2—in MCF-7 cells promotes tamoxifen resistance. We observed a sustained increase in the intracellular precursor miR-181a-2 levels in cells transfected with exogenous miR-181a-2 and demonstrated the involvement of transcription factor Snail in establishing an autoregulatory loop of miR-181a-2 that maintains cell resistance. Overall, these findings reveal a novel mechanism for reprogramming resistance-associated signaling pathways via an miR-181a-2 autoregulatory loop.
Materials and Methods
Cell lines and antiproliferative activity assay
MCF-7 breast cancer cells (ATCC, Manassas, VA, USA; HTB-22) were used. Cells were cultured in DMEM (PanEco, Moscow, Russia) supplemented with glucose (4.5 g/L) and 10% fetal bovine serum (HyClone, Marlborough, MA, USA) at 37 °C in 5% CO2. Sensitivity to tamoxifen was evaluated using the MTT assay19 with modifications from the reference.20
miRNA and wtSnail transfection
miRNA oligonucleotides were synthesized by Syntol (Moscow, Russia) and annealed in buffer (10 mM Tris-HCl pH 7.5, 50 mM NaCl, 1 mM EDTA) to obtain a 100 µM solution. Annealing was performed at 95 °C followed by slow cooling to room temperature within 1 h. Transient transfections (single and multiple) with control (scrambled) or miR-181a-2 (final concentration 50 nM) were carried out using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA). Multiple transfections (20 rounds) were performed every three days. To suppress miR-181a-2 antisense DNA oligonucleotide with LNA-modified residues was used (the sequence ASO-181a-2-3p was the following: 5’-GG[ + T]ACAGTCAACGGTCAGT[ + G]GT-3’, [ + N] – LNA modifications) synthesized by Syntol and being transfected into the cells. For ectopic expression of wild-type Snail, we used the plasmid pcDNA3-Snail-HA kindly provided by Dr. Antonio Garcia de Herreros and the corresponding empty vector.21
Generation of cells stably expressing miR-181a-2
The miR-181a-2 sequence was cloned into the lentiviral vector pLKO.1-TRC (Addgene #10878) following standard protocols. The miR-181a-2 gene was amplified from genomic DNA of healthy donor lymphocytes using primers with AgeI and EcoRI restriction sites. The amplified fragment was inserted into pLKO.1-TRC, and the construct was verified by Sanger sequencing.
Lentiviral particles were produced by transfecting HEK293FT packaging cells (Thermo Fisher Scientific) with the pLKO.1-TRC-miR-181a-2 plasmid and packaging plasmids pΔR8.2 (#12263) and pVSV-G (#8454) using GenJect-39TM (Molecta, Moscow, Russia). Viral supernatant was collected 24–28 h post-transfection and added to MCF-7 cells with 8 µg/mL Polybrene (Sigma-Aldrich). Infected cells were selected with 1 µg/mL puromycin for 4–5 days.
RNA isolation and quantitative RT-PCR
Total RNA was extracted using TRIzol reagent (Invitrogen). cDNA synthesis was performed with Advanced cDNA Synthesis Kit (Bio-Rad) from 1 µg RNA. qRT-PCR was carried out using 5X qPCRmix-HS SYBR (Evrogen) with the following conditions: 95 °C for 3 min, followed by 40 cycles of 95 °C for 15 s, 60 °C for 15 s, and 72 °C for 30 s. Human β-actin (ACTB) served as the internal control. Relative expression levels were calculated using the ΔΔCt method.22
Immunoblotting
Cell lysates were prepared as described previously in the reference.23 Proteins were separated by 10% SDS-PAGE, transferred to nitrocellulose membranes (GE Healthcare), and blocked with 5% nonfat milk (Applichem). Membranes were incubated overnight at 4 °C with primary antibodies (Cell Signaling Technology), followed by HRP-conjugated secondary antibodies (Jackson ImmunoResearch). Detection of chemiluminescence was performed using ImageQuant LAS4000 and protocol from the reference.24 Densitometry analysis for immunoblotting data was performed using ImageJ software (Wayne Rasband). The protocol for densitometry was provided by the University of Queensland with the recommendations from the reference.25
Reporter gene assay
ERE-luciferase reporter activity was measured by cotransfecting cells with ERE-luciferase and β-galactosidase plasmids (control for transfection efficiency) as described in the reference.26 The ERE-Luc plasmid was kindly provided by George Reid and Frank Gannon.27 To measure Snail trans-repressor activity using E-cadherin reporter plasmid the same approach was used, the plasmid E-cadherin-Luc was kindly provided by Prof. Antonio Garcia de Herreros.21
DNA copy number quantification
DNA copy number at the MIR181A2HG locus was quantified by qPCR using serial fourfold dilutions of genomic DNA to construct standard curves. MIR181A2HG Ct values were normalized to ACTB. Primer sequences are provided in Table 1.
Table 1.
Primer sequences
|
Gene
|
Forward primer (5’-3’)
|
Reverse primer (5’-3’)
|
TaqMan probe
|
| MIR181A2HG |
GCACAGCTGCAGGGATAGTAG |
GGCTGGAATTTCCTTCATTGT |
FAM-GCTCTCGATCCGTGGGAGGT-BHQ1 |
| MIR181A2 precursor |
TATCAGGCCAGCCTTCAGAG |
AAATCCCAAACTCACCGACA |
FAM-GACTCCAAGGAACATTCAACGC-BHQ1 |
| ACTB |
ATGTGGCCGAGGACTTTGATT |
AGTGGGGTGGCTTTTAGGATG |
Cy5-TCATTCCAAATATGAGATGCGTTGTTACAGGA-BHQ3 |
Statistical analysis
Experiments were performed in triplicate with three technical replicates. Data are expressed as mean ± SD. Mann-Whitney U test and unpaired t test were used to evaluate the experiments. Statistical significance was set at P < 0.05 (Microsoft Excel and GraphPad Prism 8).
Results
Previously, we demonstrated that prolonged treatment of estrogen-dependent MCF-7 breast cancer cells with exosomes derived from the estrogen-resistant MCF-7/T subline leads to a partial loss of (anti)estrogen sensitivity in MCF-7 cells.17 Furthermore, we showed that multiple transfections (20 sequential rounds) with one of the exosomal microRNAs, miR-181a-2, induce a similar loss of hormonal sensitivity in recipient cells, which persists for at least two months after the last transfection.28 Here, to further investigate the potential mechanism of miR-181a-2-induced acquired resistance, we analyzed the effects of multiple transfections on the expression of endogenous miR-181a-2 and associated signaling proteins.
Multiple transfections with miR-181a-2 and endogenous miR-181a-2 expression
Experiments were conducted on MCF-7 cells that underwent 20 rounds of miR-181a-2 transfection followed by maintenance in standard culture medium for at least two months. Specific PCR primers for the endogenous miR-181a-2 precursor were used to exclude contamination by exogenous miRNA mimetics and to evaluate only cellular miR-181a-2 expression. The data revealed a marked increase in endogenous miR-181a-2 precursor levels in multiply transfected cells. In contrast, a single transfection with miR-181a-2 did not affect the levels of endogenous miR-181a-2 (Figure 1a, left panel).
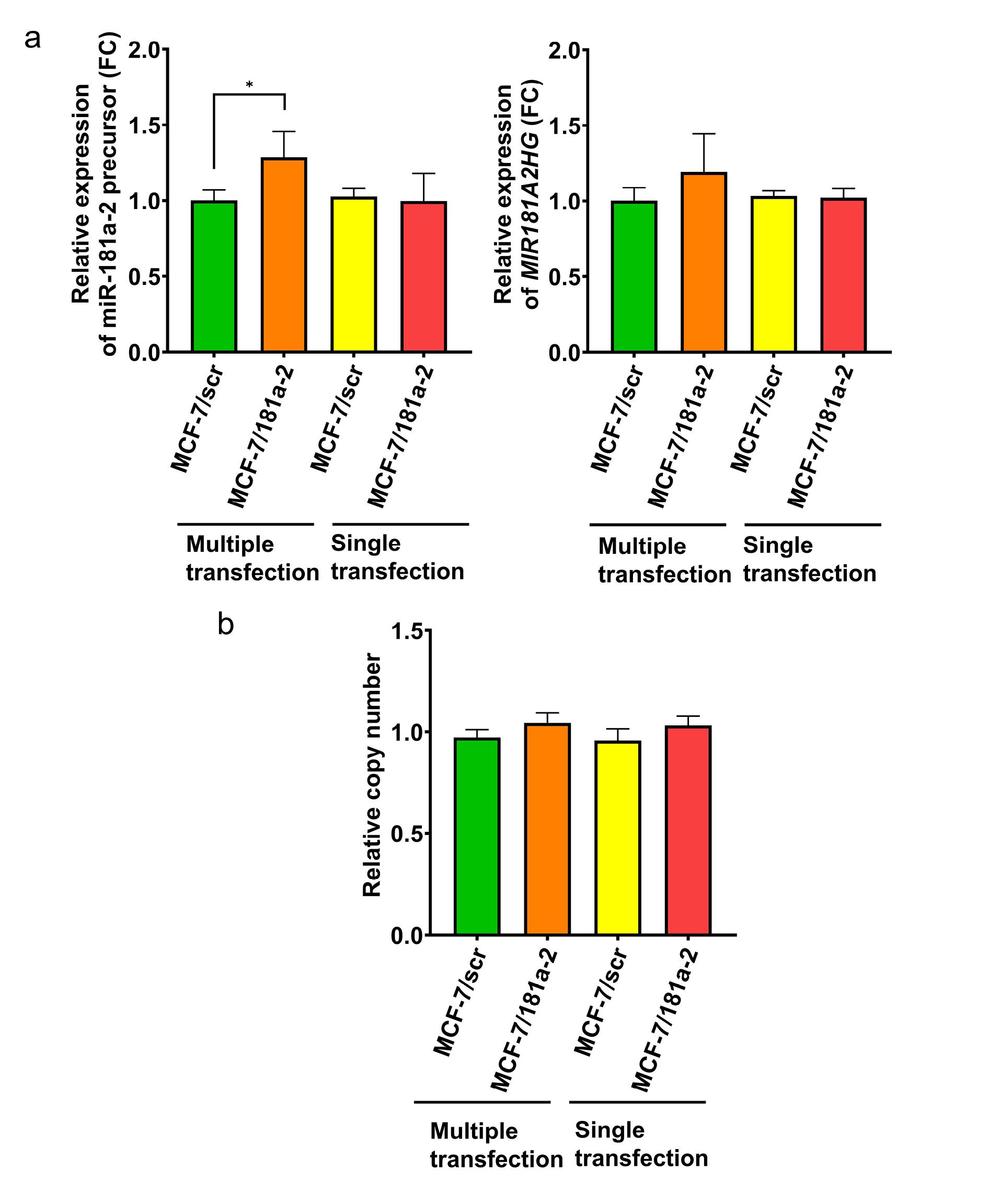
Figure 1.
(a) Expression of cellular miR-181a-2 precursor and MIR181A2HG after multiple and single miR-181a-2 transfections, *p-value = 0.0286, Mann-Whitney U test, data are presented as mean ± SD (n = 3); (b) copy number of miR-181a-2-encoded DNA in MCF-7 cells after multiple and single miR-181a-2 transfections, Mann-Whitney U test, data are presented as mean ± SD (n = 3)
.
(a) Expression of cellular miR-181a-2 precursor and MIR181A2HG after multiple and single miR-181a-2 transfections, *p-value = 0.0286, Mann-Whitney U test, data are presented as mean ± SD (n = 3); (b) copy number of miR-181a-2-encoded DNA in MCF-7 cells after multiple and single miR-181a-2 transfections, Mann-Whitney U test, data are presented as mean ± SD (n = 3)
The host gene for MIR181A2 is MIR181A2HG.29 This gene consists of two exons and one intron and encodes a long non-coding RNA; the intron harbors the MIR181A2 gene itself (Figure 2). It can therefore be assumed that miR-181a-2 is co-transcribed with MIR181A2HG. We next assessed MIR181A2HG transcription following single and multiple transfections with mature miR-181a-2. Similar to the precursor, MIR181A2HG expression increased after multiple transfections (Figure 1a, right panel).
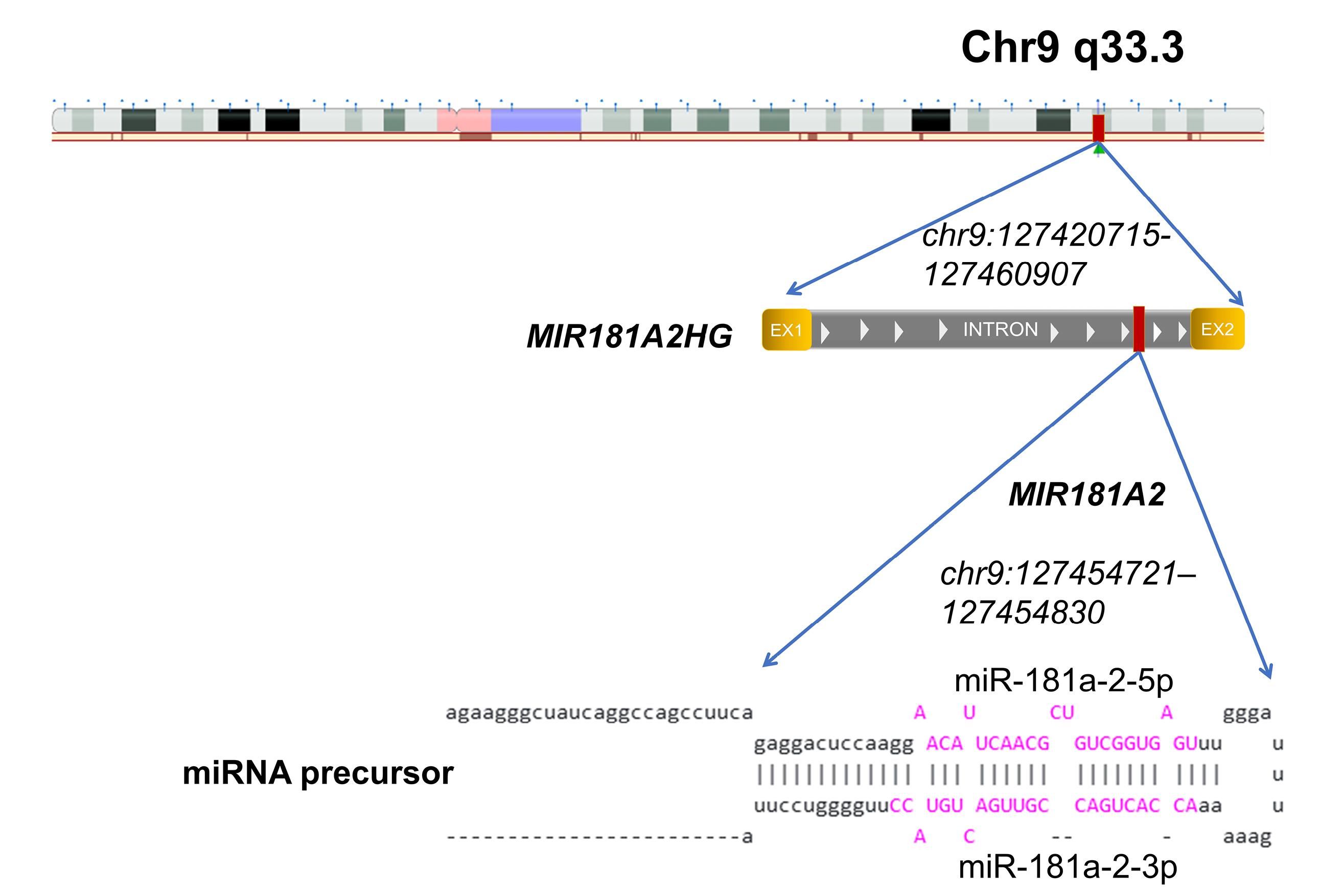
Figure 2.
Schematic representation of the MIR181A2HG and MIR181A2 genes and their genomic location. According to the UCSC Genome Browser (https://genome.ucsc.edu/, accessed 5 July 2025), MIR181A2HG is a long non-coding RNA that harbors the MIR181A2 gene. Two boxes (EX1 and EX2) indicate two exons separated by an intron of the MIR181A2HG. The predicted secondary structure of the miR-181a-2 precursor is shown in the lower panel. The mature forms miR-181a-2-5p and miR-181a-2-3p are highlighted in purple. The MIR181A2 gene is located within an intron of its host gene MIR181A2HG and is transcribed in the same direction. Genomic coordinates are provided according to the GRCh37/hg19 assembly: MIR181A2HG (chr9:127420715–127460907) and MIR181A2 (chr9:127454721–127454830)
.
Schematic representation of the MIR181A2HG and MIR181A2 genes and their genomic location. According to the UCSC Genome Browser (https://genome.ucsc.edu/, accessed 5 July 2025), MIR181A2HG is a long non-coding RNA that harbors the MIR181A2 gene. Two boxes (EX1 and EX2) indicate two exons separated by an intron of the MIR181A2HG. The predicted secondary structure of the miR-181a-2 precursor is shown in the lower panel. The mature forms miR-181a-2-5p and miR-181a-2-3p are highlighted in purple. The MIR181A2 gene is located within an intron of its host gene MIR181A2HG and is transcribed in the same direction. Genomic coordinates are provided according to the GRCh37/hg19 assembly: MIR181A2HG (chr9:127420715–127460907) and MIR181A2 (chr9:127454721–127454830)
One potential mechanism for the elevated miR-181a-2 precursor after repeated transfections with synthetic miR-181a-2 could be amplification of the genomic region encoding MIR181A2. To test this, we measured DNA copy number in the MIR181A2 locus before and after transfection. No differences were observed, excluding gene amplification as the cause of miR-181a-2 upregulation (Figure 1b).
MiR-181a-2 and estrogen receptor α signaling
As noted, a single miR-181a-2 transfection induces only transient estrogen resistance, whereas 20 transfections confer irreversible tamoxifen resistance lasting at least two months after the final transfection.28 To further investigate the effects of continuous miR-181a-2 uptake on ERα signaling, we infected MCF-7 cells with a lentiviral construct expressing miR-181a-2. Similar to multiple transfections, lentivirus-infected cells displayed stably increased levels of both the miR-181a-2 precursor and MIR181A2HG RNA (Figure 3a). Tamoxifen sensitivity assays revealed partial resistance in the transfected cells (Figure 3b). Moreover, miR-181a-2-overexpressing cells showed irreversible suppression of ERα signaling, including inhibition of ERα expression and transcriptional activity and downregulation of ERα-dependent proteins GREB1 and PR (Figure 3c, d). These results confirm a direct link between miR-181a-2 overexpression and ERα pathway suppression.
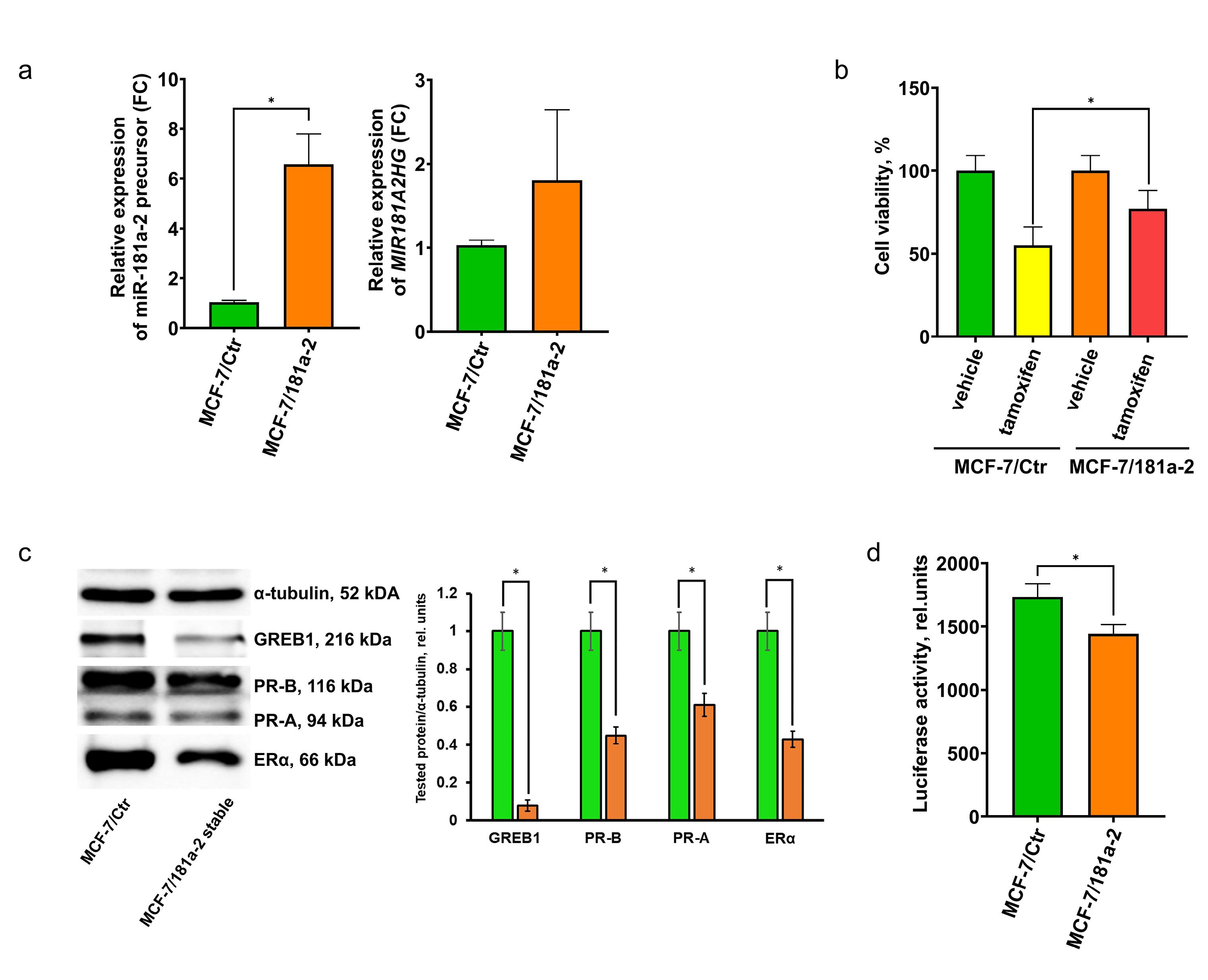
Figure 3.
(a) Expression of cellular miR-181a-2 precursor and MIR181A2HG in MCF-7 cells stably infected with miR-181a-2, *P value = 0.0032, unpaired t-test, data are presented as mean ± SD (n = 3); (b) sensitivity of MCF-7/Ctr and MCF-7/miR-181a-2 cells to tamoxifen, assessed by MTT assay, *P value = 0.0317, unpaired t test, data are presented as mean ± SD (n = 3); (c) Western blot analysis of ERα-dependent proteins in MCF-7/Ctr and MCF-7/miR-181a-2 cells. The blot shows results representative of three independent experiments; t-test, *P= 0.0012 for GREB1, 0.013 for PR-B, 0.015 for PR-A, 0.031 for erα (d) reporter assay of ERα transcriptional activity in MCF-7/Ctr and MCF-7/miR-181a-2. Relative luciferase activity was calculated as the ratio of luciferase to β-galactosidase activity (arbitrary units); *P value = 0.016, unpaired t test, data are presented as mean ± SD (n = 3)
.
(a) Expression of cellular miR-181a-2 precursor and MIR181A2HG in MCF-7 cells stably infected with miR-181a-2, *P value = 0.0032, unpaired t-test, data are presented as mean ± SD (n = 3); (b) sensitivity of MCF-7/Ctr and MCF-7/miR-181a-2 cells to tamoxifen, assessed by MTT assay, *P value = 0.0317, unpaired t test, data are presented as mean ± SD (n = 3); (c) Western blot analysis of ERα-dependent proteins in MCF-7/Ctr and MCF-7/miR-181a-2 cells. The blot shows results representative of three independent experiments; t-test, *P= 0.0012 for GREB1, 0.013 for PR-B, 0.015 for PR-A, 0.031 for erα (d) reporter assay of ERα transcriptional activity in MCF-7/Ctr and MCF-7/miR-181a-2. Relative luciferase activity was calculated as the ratio of luciferase to β-galactosidase activity (arbitrary units); *P value = 0.016, unpaired t test, data are presented as mean ± SD (n = 3)
MiR-181a-2 and signaling protein expression
Analysis of growth-related proteins in miR-181a-2-transfected cells revealed stable activation of CDK6, cyclin D1, and Snail, whereas major effectors of mTOR and PI3K/Akt pathways were unaffected (Figure 4a).
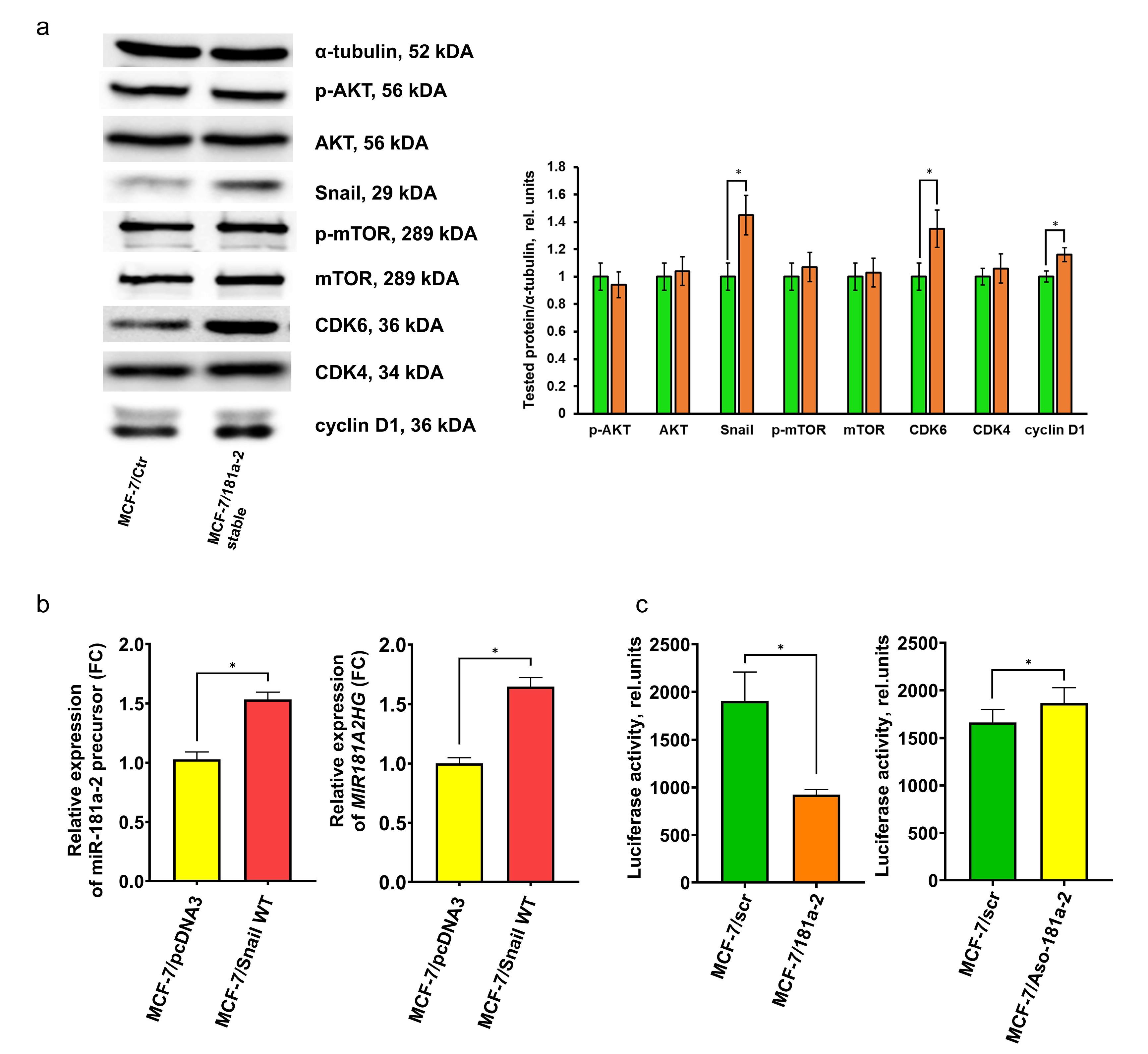
Figure 4.
(a) Western blot analysis of growth-related proteins in MCF-7/Ctr and MCF-7/miR-181a-2 cells. The blot shows results representative of three independent experiments; t-test, *P= 0.034 for Snail, 0.035 for CDK6, 0.045 for cyclin D1 (b) expression of cellular miR-181a-2 precursor (left panel, *P value = 0.0005, unpaired t test) and MIR181A2HG (right panel, *P value = 0.0002, unpaired t test) in MCF-7 cells following wtSnail transfection, data are presented as mean ± SD (n = 3); (c) reporter assay of E-cadherin transcriptional activity in the cells after transfection of scrambled miR/miR-181a-2 (left panel, *P value = 0.0007, unpaired t test) and after transfection of antisense (Aso) DNA oligonucleotides scrambled/Aso-miR-181a-2 (right panel, *P value = 0.0398, unpaired t test), data are presented as mean ± SD (n = 3)
.
(a) Western blot analysis of growth-related proteins in MCF-7/Ctr and MCF-7/miR-181a-2 cells. The blot shows results representative of three independent experiments; t-test, *P= 0.034 for Snail, 0.035 for CDK6, 0.045 for cyclin D1 (b) expression of cellular miR-181a-2 precursor (left panel, *P value = 0.0005, unpaired t test) and MIR181A2HG (right panel, *P value = 0.0002, unpaired t test) in MCF-7 cells following wtSnail transfection, data are presented as mean ± SD (n = 3); (c) reporter assay of E-cadherin transcriptional activity in the cells after transfection of scrambled miR/miR-181a-2 (left panel, *P value = 0.0007, unpaired t test) and after transfection of antisense (Aso) DNA oligonucleotides scrambled/Aso-miR-181a-2 (right panel, *P value = 0.0398, unpaired t test), data are presented as mean ± SD (n = 3)
To identify factors responsible for sustained endogenous miR-181a-2 overexpression, we examined the relationship between miR-181a-2 and Snail, a resistance-associated protein.30-32 We observed direct accumulation of endogenous miR-181a-2 in Snail-overexpressing cells, highlighting Snail’s involvement in the positive regulation and maintenance of miR-181a-2 levels (Figure 4b). Furthermore, miR-181a-2 transfection resulted in the activation of Snail trans-repressor activity (Figure 4c, left panel), whereas knockdown of miR-181a-2 using antisense (ASO) DNA oligonucleotides showed a slight tendency toward Snail suppression (Figure 4c, right panel). The modest effect observed in the latter case may be due to the relatively low number of ASO copies compared to endogenous cellular miR-181a-2 levels. Taken together, these findings suggest the possible formation of an autoregulatory loop between Snail and miR-181a-2, which may be responsible for maintenance of this signaling pathway.
Overall, we uncovered a phenomenon of reciprocal activation: continuous uptake of exogenous miR-181a-2 mimetics induces sustained endogenous miR-181a-2 upregulation, forming an autoregulatory loop that contributes to hormonal resistance. This loop appears to be supported, at least in part, by Snail activation.
Discussion
Breast cancer is predominantly hormone-dependent, with estrogen receptors expressed in over 70% of cases, making them critical therapeutic targets. Hormone therapy—aimed at ERα inactivation or depletion of endogenous estrogens—is limited by the emergence of drug resistance, which can be either primary (present at diagnosis) or acquired (developing during therapy). Primary resistance is often associated with gene mutations disrupting hormonal signaling (e.g., receptor repression) or activating hormone-independent growth pathways (e.g., tyrosine kinase cascades). Acquired resistance is usually epigenomic in nature, with microRNAs playing key roles by directly suppressing specific genes or indirectly modulating DNA methylation and acetylation.
Numerous microRNAs have been implicated in the development of hormonal resistance in tumors. These include ERα-negative regulators such as miR-342,33 Let-7b/Let-7i,34 and miR-1280.35 Additionally, miRNAs targeting ERα coactivators/corepressors are involved: miR-17-5p regulates SRC-336; miR-10 targets the nuclear corepressor NCOR2;16 miR-451 regulates HER2, EGFR, and MAPK signaling12; and miR-101 influences Akt signaling in resistant cells.13 Among tumor suppressors, PTEN is particularly notable as a frequent miRNA target linked to hormonal resistance.37-39 Several of these miRNAs are known to be exosome-transported, supporting their role in resistance transfer.40,41
Recently, increasing attention has focused on exosome-mediated resistance formation. Exosomes—microvesicles secreted by cells into the extracellular environment—can be taken up by recipient cells, transferring regulatory miRNAs that affect hormone signaling.12,13,16,36 These miRNAs can modulate proliferation and expression of key proteins.42-44 However, the duration of miRNA influence on signaling and involvement of proteins not directly targeted by specific miRNAs remain unclear. Given that miRNAs are delivered in complex mixtures containing hundreds of species, it is crucial to identify specific miRNAs and link them to defined cellular signaling changes.
In our previous work with estrogen-dependent MCF-7 cells and tamoxifen-resistant MCF-7/T cells, we demonstrated that exosomes from resistant cells can induce hormonal resistance in parental cells. Profiling of microRNAs in “resistant” exosomes revealed overexpression of multiple ERα-targeting miRNAs, including miR-181a-2. We subsequently confirmed miR-181a-2 as a key driver of tamoxifen resistance.28 These findings are consistent with other studies linking miR-181 to drug resistance in various models.45-47 Nonetheless, the mechanisms underlying long-term maintenance of resistance in newly generated resistant cells remain unclear.
Here, for the first time, we describe hyperexpression of endogenous miR-181a-2 precursor in MCF-7 cells following multiple (20 rounds) transfections with exogenous miR-181a-2 mimetics. Elevated precursor levels persisted for at least two months post-transfection. Multiply transfected cells exhibited partial hormonal insensitivity, suppression of estrogen signaling, DNMT3A inhibition, and increased expression of growth-related proteins, consistent with known properties of miR-181.14,47-50
Among these proteins, we identified Snail—an EMT regulator and indirect miR-181 target. Our results are consistent with the data of other researchers who have demonstrated the increase in Snail level in the miR-181-overexpressed cells.51-53 Partially, the recent observations have shown the ability of miR-181a, along with TMBIM6 protein, to increase Snail level through the activation of ERK pathway in breast cancer cells.51
We and others have previously shown that Snail contributes to resistance to hormonal drugs.30-32 Consistent with this, we observed an association between Snail overexpression and elevated miR-181a-2 precursor levels, implicating Snail as a key mediator of miR-181a-2-induced resistance. Finally, we have demonstrated the formation of autoregulatory loop between Snail and mir-181a-2 that may be responsible for cell resistance phenotype.
Conclusion
We propose a mechanism of cancer cell resistance based on an autoregulatory loop sustaining high levels of resistance-associated factors, such as miR-181a-2. Snail plays a central role in this loop: it both activates miR-181a-2 and is itself a target of miR-181a-2. The precise mediators transmitting signals from Snail to miR-181a-2, the mechanisms by which this loop is preserved during cell division, and its role in biochemical imprinting and broader phenotypic adaptation of cancer cells remain open questions for future investigation.
Competing Interests
All authors declared that there are no conflicts of interest.
Consent for publication
Not applicable.
Data Availability Statement
The datasets of the current study are available from the corresponding author upon request.
Ethical Approval
Not applicable.
Acknowledgements
The authors thank George Reid and Frank Gannon for the shared plasmids. We thank Alvina Khamidullina and the Center for Precision Genome Editing and Genetic Technologies for Biomedicine, Institute of Gene Biology, Russian Academy of Sciences, for providing the equipment of research facilities (gel imaging system).
References
- Gautam S, Maurya R, Vikal A, Patel P, Thakur S, Singh A. Understanding drug resistance in breast cancer: mechanisms and emerging therapeutic strategies. Med Drug Discov 2025; 26:100210. doi: 10.1016/j.medidd.2025.100210 [Crossref] [ Google Scholar]
- Scherbakov AM, Krasil’nikov MA, Kushlinskii NE. Molecular mechanisms of hormone resistance of breast cancer. Bull Exp Biol Med 2013; 155(3):384-95. doi: 10.1007/s10517-013-2160-y [Crossref] [ Google Scholar]
- Andreeva OE, Sorokin DV, Vinokurova SV, Kopnin PB, Elkina NV, Katargin AN. Breast cancer cell resistance to hormonal and targeted therapeutics is correlated with the inactivation of the NR6A1 axis. Cancer Drug Resist 2024; 7:48. doi: 10.20517/cdr.2024.69 [Crossref] [ Google Scholar]
- Hartkopf AD, Grischke EM, Brucker SY. Endocrine-resistant breast cancer: mechanisms and treatment. Breast Care (Basel) 2020; 15(4):347-54. doi: 10.1159/000508675 [Crossref] [ Google Scholar]
- Khan A, Sisodiya S, Aftab M, Tanwar P, Hussain S, Gupta V. Mechanisms and therapeutic strategies for endocrine resistance in breast cancer: a comprehensive review and meta-analysis. Cancers (Basel) 2025; 17(10):1653. doi: 10.3390/cancers17101653 [Crossref] [ Google Scholar]
- Muluhngwi P, Klinge CM. Roles for miRNAs in endocrine resistance in breast cancer. EndocrRelat Cancer 2015; 22(5):R279-300. doi: 10.1530/erc-15-0355 [Crossref] [ Google Scholar]
- Leivonen SK, Mäkelä R, Ostling P, Kohonen P, Haapa-Paananen S, Kleivi K. Protein lysate microarray analysis to identify microRNAs regulating estrogen receptor signaling in breast cancer cell lines. Oncogene 2009; 28(44):3926-36. doi: 10.1038/onc.2009.241 [Crossref] [ Google Scholar]
- Guttilla IK, Adams BD, White BA. ERα, microRNAs, and the epithelial-mesenchymal transition in breast cancer. Trends Endocrinol Metab 2012; 23(2):73-82. doi: 10.1016/j.tem.2011.12.001 [Crossref] [ Google Scholar]
- Yoshimoto N, Toyama T, Takahashi S, Sugiura H, Endo Y, Iwasa M. Distinct expressions of microRNAs that directly target estrogen receptor α in human breast cancer. Breast Cancer Res Treat 2011; 130(1):331-9. doi: 10.1007/s10549-011-1672-2 [Crossref] [ Google Scholar]
- Sheng B, Yuan Y, Liu X, Zhang Y, Liu H, Shen X. Protective effect of estrogen against intervertebral disc degeneration is attenuated by miR-221 through targeting estrogen receptor α. Acta BiochimBiophys Sin (Shanghai) 2018; 50(4):345-54. doi: 10.1093/abbs/gmy017 [Crossref] [ Google Scholar]
- Mansoori B, Mohammadi A, Gjerstorff MF, Shirjang S, Asadzadeh Z, Khaze V. miR-142-3p is a tumor suppressor that inhibits estrogen receptor expression in ER-positive breast cancer. J Cell Physiol 2019; 234(9):16043-53. doi: 10.1002/jcp.28263 [Crossref] [ Google Scholar]
- Bergamaschi A, Katzenellenbogen BS. Tamoxifen downregulation of miR-451 increases 14-3-3ζ and promotes breast cancer cell survival and endocrine resistance. Oncogene 2012; 31(1):39-47. doi: 10.1038/onc.2011.223 [Crossref] [ Google Scholar]
- Sachdeva M, Wu H, Ru P, Hwang L, Trieu V, Mo YY. MicroRNA-101-mediated Akt activation and estrogen-independent growth. Oncogene 2011; 30(7):822-31. doi: 10.1038/onc.2010.463 [Crossref] [ Google Scholar]
- Strotbek M, Schmid S, Sánchez-González I, Boerries M, Busch H, Olayioye MA. miR-181 elevates Akt signaling by co-targeting PHLPP2 and INPP4B phosphatases in luminal breast cancer. Int J Cancer 2017; 140(10):2310-20. doi: 10.1002/ijc.30661 [Crossref] [ Google Scholar]
- Jian B, Li Z, Xiao D, He G, Bai L, Yang Q. Downregulation of microRNA-193-3p inhibits tumor proliferation migration and chemoresistance in human gastric cancer by regulating PTEN gene. Tumour Biol 2016; 37(7):8941-9. doi: 10.1007/s13277-015-4727-x [Crossref] [ Google Scholar]
- Foley NH, Bray I, Watters KM, Das S, Bryan K, Bernas T. MicroRNAs 10a and 10b are potent inducers of neuroblastoma cell differentiation through targeting of nuclear receptor corepressor 2. Cell Death Differ 2011; 18(7):1089-98. doi: 10.1038/cdd.2010.172 [Crossref] [ Google Scholar]
- Semina SE, Scherbakov AM, Vnukova AA, Bagrov DV, Evtushenko EG, Safronova VM. Exosome-mediated transfer of cancer cell resistance to antiestrogen drugs. Molecules 2018; 23(4):829. doi: 10.3390/molecules23040829 [Crossref] [ Google Scholar]
- Semina SE, Scherbakov AM, Kovalev SV, Shevchenko VE, Krasil’nikov MA. Horizontal transfer of tamoxifen resistance in MCF-7 cell derivates: proteome study. Cancer Invest 2017; 35(8):506-18. doi: 10.1080/07357907.2017.1368081 [Crossref] [ Google Scholar]
- Iselt M, Holtei W, Hilgard P. The tetrazolium dye assay for rapid in vitro assessment of cytotoxicity. Arzneimittelforschung 1989; 39(7):747-9. [ Google Scholar]
- Scherbakov AM, Komkov AV, Komendantova AS, Yastrebova MA, Andreeva OE, Shirinian VZ. Steroidal pyrimidines and dihydrotriazines as novel classes of anticancer agents against hormone-dependent breast cancer cells. Front Pharmacol 2017; 8:979. doi: 10.3389/fphar.2017.00979 [Crossref] [ Google Scholar]
- Dave N, Guaita-Esteruelas S, Gutarra S, Frias À, Beltran M, Peiró S. Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J Biol Chem 2011; 286(14):12024-32. doi: 10.1074/jbc.M110.168625 [Crossref] [ Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001; 25(4):402-8. doi: 10.1006/meth.2001.1262 [Crossref] [ Google Scholar]
- Kuznetsov YV, Levina IS, Scherbakov AM, Andreeva OE, Dmitrenok AS, Malyshev OR. 3,20-Dihydroxy-13α-19-norpregna-1,3,5(10)-trienes Synthesis, structures, and cytotoxic, estrogenic, and antiestrogenic effects. Steroids 2018; 137:1-13. doi: 10.1016/j.steroids.2018.07.007 [Crossref] [ Google Scholar]
- Mruk DD, Cheng CY. Enhanced chemiluminescence (ECL) for routine immunoblotting: an inexpensive alternative to commercially available kits. Spermatogenesis 2011; 1(2):121-2. doi: 10.4161/spmg.1.2.16606 [Crossref] [ Google Scholar]
- Taylor SC, Berkelman T, Yadav G, Hammond M. A defined methodology for reliable quantification of Western blot data. Mol Biotechnol 2013; 55(3):217-26. doi: 10.1007/s12033-013-9672-6 [Crossref] [ Google Scholar]
- Ilovaisky AI, Scherbakov AM, Merkulova VM, Chernoburova EI, Shchetinina MA, Andreeva OE. Secosteroid–quinoline hybrids as new anticancer agents. J Steroid Biochem Mol Biol 2023; 228:106245. doi: 10.1016/j.jsbmb.2022.106245 [Crossref] [ Google Scholar]
- Reid G, Hübner MR, Métivier R, Brand H, Denger S, Manu D. Cyclic, proteasome-mediated turnover of unliganded and liganded ERα on responsive promoters is an integral feature of estrogen signaling. Mol Cell 2003; 11(3):695-707. doi: 10.1016/s1097-2765(03)00090-x [Crossref] [ Google Scholar]
- Andreeva OE, Sorokin DV, Mikhaevich EI, Bure IV, Shchegolev YY, Nemtsova MV. Towards unravelling the role of ERα-targeting miRNAs in the exosome-mediated transferring of the hormone resistance. Molecules 2021; 26(21):6661. doi: 10.3390/molecules26216661 [Crossref] [ Google Scholar]
- Xu Y, Chen J, Yang Z, Xu L. Identification of RNA expression profiles in thyroid cancer to construct a competing endogenous RNA (ceRNA) network of mRNAs, long noncoding RNAs (lncRNAs), and microRNAs (miRNAs). Med Sci Monit 2019; 25:1140-54. doi: 10.12659/msm.912450 [Crossref] [ Google Scholar]
- Scherbakov AM, Andreeva OE, Shatskaya VA, Krasil’nikov MA. The relationships between snail1 and estrogen receptor signaling in breast cancer cells. J Cell Biochem 2012; 113(6):2147-55. doi: 10.1002/jcb.24087 [Crossref] [ Google Scholar]
- Scherbakov AM, Sorokin DV, Tatarskiy VV Jr, Prokhorov NS, Semina SE, Berstein LM. The phenomenon of acquired resistance to metformin in breast cancer cells: the interaction of growth pathways and estrogen receptor signaling. IUBMB Life 2016; 68(4):281-92. doi: 10.1002/iub.1481 [Crossref] [ Google Scholar]
- Takeda T, Tsubaki M, Matsuda T, Kimura A, Jinushi M, Obana T. EGFR inhibition reverses epithelial-mesenchymal transition, and decreases tamoxifen resistance via Snail and Twist downregulation in breast cancer cells. Oncol Rep 2022; 47(6):109. doi: 10.3892/or.2022.8320 [Crossref] [ Google Scholar]
- He YJ, Wu JZ, Ji MH, Ma T, Qiao EQ, Ma R. miR-342 is associated with estrogen receptor-α expression and response to tamoxifen in breast cancer. Exp Ther Med 2013; 5(3):813-8. doi: 10.3892/etm.2013.915 [Crossref] [ Google Scholar]
- Zhao Y, Deng C, Lu W, Xiao J, Ma D, Guo M. let-7 microRNAs induce tamoxifen sensitivity by downregulation of estrogen receptor α signaling in breast cancer. Mol Med 2011; 17(11-12):1233-41. doi: 10.2119/molmed.2010.00225 [Crossref] [ Google Scholar]
- Meng D, Li Z, Ma X, Fu L, Qin G. MicroRNA-1280 modulates cell growth and invasion of thyroid carcinoma through targeting estrogen receptor α. Cell Mol Biol (Noisy-le-grand) 2016; 62(3):1-6. [ Google Scholar]
- Hossain A, Kuo MT, Saunders GF. Mir-17-5p regulates breast cancer cell proliferation by inhibiting translation of AIB1 mRNA. Mol Cell Biol 2006; 26(21):8191-201. doi: 10.1128/mcb.00242-06 [Crossref] [ Google Scholar]
- Chen WX, Liu XM, Lv MM, Chen L, Zhao JH, Zhong SL. Exosomes from drug-resistant breast cancer cells transmit chemoresistance by a horizontal transfer of microRNAs. PLoS One 2014; 9(4):e95240. doi: 10.1371/journal.pone.0095240 [Crossref] [ Google Scholar]
- Phuong NT, Kim SK, Im JH, Yang JW, Choi MC, Lim SC. Induction of methionine adenosyltransferase 2A in tamoxifen-resistant breast cancer cells. Oncotarget 2016; 7(12):13902-16. doi: 10.18632/oncotarget.5298 [Crossref] [ Google Scholar]
- Yu X, Li R, Shi W, Jiang T, Wang Y, Li C. Silencing of microRNA-21 confers the sensitivity to tamoxifen and fulvestrant by enhancing autophagic cell death through inhibition of the PI3K-AKT-mTOR pathway in breast cancer cells. Biomed Pharmacother 2016; 77:37-44. doi: 10.1016/j.biopha.2015.11.005 [Crossref] [ Google Scholar]
- Melo SA, Sugimoto H, O’Connell JT, Kato N, Villanueva A, Vidal A. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 2014; 26(5):707-21. doi: 10.1016/j.ccell.2014.09.005 [Crossref] [ Google Scholar]
- Asgari R, Rezaie J. Differential expression of serum exosomal miRNAs in breast cancer patients and healthy controls. Adv Pharm Bull 2022; 12(4):858-62. doi: 10.34172/apb.2022.088 [Crossref] [ Google Scholar]
- Mozammel N, Baghbani E, Amini M, Jodeiry Zaer S, Baghay Esfandyari Y, Tohidast M. The simultaneous effects of miR-145-5p and hsa-let-7a-3p on colorectal tumorigenesis: in vitro evidence. Adv Pharm Bull 2024; 14(1):231-40. doi: 10.34172/apb.2024.004 [Crossref] [ Google Scholar]
- Jodeiry Zaer S, Aghamaali M, Amini M, Doustvandi MA, Hosseini SS, Baradaran B. Cooperatively inhibition effect of miR-143-5p and miR-145-5p in tumorigenesis of glioblastoma cells through modulating AKT signaling pathway. Bioimpacts 2024; 14(3):29913. doi: 10.34172/bi.2023.29913 [Crossref] [ Google Scholar]
- Bilan F, Amini M, Doustvandi MA, Tohidast M, Baghbanzadeh A, Hosseini SS. Simultaneous suppression of miR-21 and restoration of miR-145 in gastric cancer cells; a promising strategy for inhibition of cell proliferation and migration. Bioimpacts 2024; 14(2):27764. doi: 10.34172/bi.2023.27764 [Crossref] [ Google Scholar]
- Suresh Babu V, Bisht A, Mallipatna A, Sa D, Dudeja G, Kannan R. Enhanced epithelial-to-mesenchymal transition and chemoresistance in advanced retinoblastoma tumors is driven by miR-181a. Cancers (Basel) 2022; 14(20):5124. doi: 10.3390/cancers14205124 [Crossref] [ Google Scholar]
- Gao L, Wang G, Liu WN, Kinser H, Franco HL, Mendelson CR. Reciprocal feedback between miR-181a and E2/ERα in myometrium enhances inflammation leading to labor. J Clin Endocrinol Metab 2016; 101(10):3646-56. doi: 10.1210/jc.2016-2078 [Crossref] [ Google Scholar]
- Zheng Y, Lv X, Wang X, Wang B, Shao X, Huang Y. MiR-181b promotes chemoresistance in breast cancer by regulating Bim expression. Oncol Rep 2016; 35(2):683-90. doi: 10.3892/or.2015.4417 [Crossref] [ Google Scholar]
- Zhang Y, Guan Y, Zheng X, Li C. Hypoxia-induced miR-181a-5p up-regulation reduces epirubicin sensitivity in breast cancer cells through inhibiting EPDR1/TRPC1 to activate PI3K/AKT signaling pathway. BMC Cancer 2024; 24(1):167. doi: 10.1186/s12885-024-11906-6 [Crossref] [ Google Scholar]
- Henao-Mejia J, Williams A, Goff LA, Staron M, Licona-Limón P, Kaech SM. The microRNA miR-181 is a critical cellular metabolic rheostat essential for NKT cell ontogenesis and lymphocyte development and homeostasis. Immunity 2013; 38(5):984-97. doi: 10.1016/j.immuni.2013.02.021 [Crossref] [ Google Scholar]
- Liu J, Xu D, Wang Q, Zheng D, Jiang X, Xu L. LPS induced miR-181a promotes pancreatic cancer cell migration via targeting PTEN and MAP2K4. Dig Dis Sci 2014; 59(7):1452-60. doi: 10.1007/s10620-014-3049-y [Crossref] [ Google Scholar]
- Shin Y, Choi HY, Kwak Y, Yang GM, Jeong Y, Jeon TI. TMBIM6-mediated miR-181a expression regulates breast cancer cell migration and invasion via the MAPK/ERK signaling pathway. J Cancer 2023; 14(4):554-72. doi: 10.7150/jca.81600 [Crossref] [ Google Scholar]
- Hu W, Yan F, Ru Y, Xia M, Yan G, Zhang M. MIIP inhibits EMT and cell invasion in prostate cancer through miR-181a/b-5p-KLF17 axis. Am J Cancer Res 2020; 10(2):630-47. [ Google Scholar]
- Chitsazzadeh V, Nguyen TN, de Mingo Pulido A, Bittencourt BB, Du L, Adelmann CH, et al. miR-181a promotes multiple protumorigenic functions by targeting TGFβR3. J Invest Dermatol 2022;142(7):1956-65.e2. doi: 10.1016/j.jid.2021.09.040.