Advanced pharmaceutical bulletin. 10(3):423-429.
doi: 10.34172/apb.2020.051
Research Article
Design, Synthesis and Biological Evaluation of Novel Piperazinone Derivatives as Cytotoxic Agents
Saeed Ghasemi 1  , Simin Sharifi 2, Javid Shahbazi Mojarrad 3, *
, Simin Sharifi 2, Javid Shahbazi Mojarrad 3, *
Author information:
1Department of Medicinal Chemistry, School of Pharmacy, Guilan University of Medical Sciences, Rasht, Iran.
2Dental and Periodontal Research Center, Tabriz University of Medical Sciences, Tabriz, Iran.
3Department of Medicinal Chemistry, Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran.
Abstract
Purpose:
In this study, a series of piperazin-2-one derivatives were prepared through bioisosteric substitution of the imidazole ring of L-778,123 (imidazole-containing FTase inhibitor) and rearrangement of groups based on the tipifarnib structure. Final compounds were evaluated for their cytotoxic activities on cancer and normal cell lines by MTT assay.
Methods: Methyl α-bromophenylacetic acid and 1-(3-chlorophenyl) piperazin-2-one were synthesized using previously described methods. Methyl 2-(4-chlorophenyl)-2-(4-(3- chlorophenyl)-3-oxopiperazin-1-yl) acetate was prepared by reaction between these two compounds in presence of potassium carbonate. Finally, methoxy group of ester was substituted by various amines such as guanidine, thiourea, urea and hydrazide. The synthesized compounds were tested for their cytotoxicity against colon cancer (HT-29) and lung cancer (A549) cell lines as well as MRC-5 (normal fetal lung fibroblasts) cells as a healthy cell line using MTT colorimetric assay method.
Results: Replacement of imidazole moiety with guanidine, thiourea, and hydrazide could increase cytotoxicity toward all three cell lines. Some substituents, such as amine, urea, and hydroxylamine exhibited significant cytotoxicity (<500 µM) but lower than L-778,123 as standard compound. Hydroxyl and methoxy substituents did not show significant cytotoxicity. Imidazole substituent group revealed cytotoxicity similar to L-778,123 All compounds showed lower cytotoxic activity against normal cell lines compared with cancer cell lines.
Conclusion: It seems the electron density of substituted groups and rearrangement of groups may significantly increase cytotoxic activity
Keywords: Anticancer activity, Bioisosteric replacement, Piperazinone, Chemical synthesis
Copyright and License Information
© 2020 The Authors.
This is an Open Access article distributed under the terms of the Creative Commons Attribution (CC BY), which permits unrestricted use, distribution, and reproduction in any medium, as long as the original authors and source are cited. No permission is required from the authors or the publishers.
Introduction
In developed countries, cancer has changed into one of the most important causes of death. Because of the complications of existing agents such as drug resistance and toxicities, the introduction of novel anticancer compounds is essential.1,2+
Studies have demonstrated that 30% of human cancers occur as a result of the mutation of ras genes.3 Ras proteins activate signal transduction pathways with an essential role in cell growth. The post-translational modification should be done for activation by several sequential enzymatic steps. Farnesylation of Ras protein can be interrupted by farnesyltransferase inhibitors (FTIs) and cause suppression of the tumor cells growth that depends on Ras.4,5 Thus, the research for the development of the cancer treatment by novel FTIs has recently attracted a great deal of attention. Several potent non-thiol FTIs such as lonafarnib, L-778,123, BMS-214662, and tipifarnib have been introduced which can be used for hematological cancers and solid tumors therapy (Figure 1).5-9
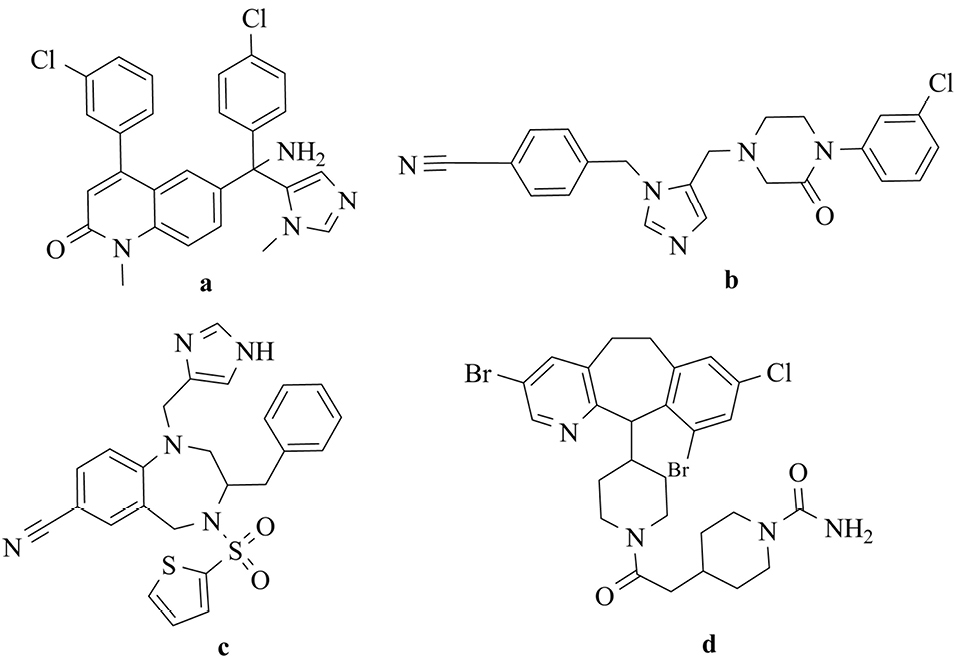
Figure 1.
Structures of FTIs: (a) L-778,123 (b)Tipifarnib (c)BMS-214662 (d)Lonafarnib.
.
Structures of FTIs: (a) L-778,123 (b)Tipifarnib (c)BMS-214662 (d)Lonafarnib.
Imidazole ring has a crucial role in the interaction of these groups with the enzyme. Researchers have shown that although the imidazole substitution with other heterocyclic rings such as pyridine in FTIs can improve the cytotoxicity, it can decrease inhibitory activity on human farnesyltransferase. Besides, studies of cancerous cell lines have suggested that more than 70% of the cancer cells without mutation in Ras proteins can be sensitive to FTIs.4,5 These results revealed the existence of extra mechanisms apart from farnesyltransferase enzyme inhibition.9-13
Also, some groups such as semicarbazide, guanidine, thiourea, and urea derivatives exhibit potent cytotoxic activity with diverse mechanisms.14-18
In this research, the synthesis and cytotoxic activity profile of new 1-(3-chlorophenyl) 2-piperazinone compounds containing imidazole moiety and bioisosteres of imidazole such as guanidine, thiourea, etc. were reported. The study also explores the structure of compounds rearranged based on the tipifarnibe structure as potent FTIs. The novel compounds are investigated on normal human lung cells as well as two human cancerous cells including MRC-5 (normal fetal lung fibroblasts) and HT29 (human colonic adenocarcinoma cells) and A549 (adenocarcinoma human alveolar basal epithelial cells) cells.
Materials and Methods
Chemistry
Reagents and solvents were bought from Merck and Sigma Aldrich. An Electrothermal-9100 melting point apparatus was used for the melting point determination and are uncorrected. Shimadzu 4300 spectrophotometer (potassium bromide dicks) was used for recording the IR spectra. 1HNMR and 13CNMR spectra were acquired using a Bruker spectrometer (FT-500 and 400 MHz) with tetramethylsilane (TMS) as the internal standard. The elemental analyses were done by the CHN-O elemental analyzer by GmbH-Germany for contents of C, H, and N (The results are reported within ±0.4% of the calculated values). The mass data were recorded on a LC-MS (Agilent 6410) at 70 eV. Merck silica gel 60 F254 plates were used for analytical thin-layer chromatography (TLC). Particle size 0.06-0.20 mm (Merck) was used for column chromatography.
α-Bromo(4-chlorophenyl)acetic acid(1)
A solution of 4-chlorophenylacetic acid (10 g, 58.62 mmol), phosphorus trichloride (0.52 g, 4.16 mmol), and bromine (9.99 g, 62.43 mmol) was refluxed in 750 ml of benzene for three days. After cooling the solution to 25°C, it was filtered. Obtained brown oil by the solvent evaporation under reduced pressure was crystallized from ligroin. Finally, the crystals were filtered, washed by cool ligroin, and then dried.
Yield: 60%; mp = 98-100°C; IR (KBr, cm-1) νmax: 2400-3400(OH), 1720(C=O), 670 (C-Br). 1H NMR (CDCl3, 400 MHz): δ ppm 7.58 (d, 2H, J = 8.5 Hz, phenyl), 7.47 (d, 2H, J = 8.5 Hz, phenyl), 5.80 (s, 1H, CHBr).
Methyl α-Bromophenylacetic acid (2)
α-Bromo(4-chlorophenyl) acetic acid (10g, 40.08mmol) obtained from the previous step and concentrated sulfuric acid (5 g) were refluxed in methanol (40 mL) for 4 hours. The solvent was evaporated by reduced pressure. Dichloromethane (50 mL) and sodium bicarbonate (NaHCO3) solution (50 mL) were added to the precipitate. After drying the dichloromethane phase by sodium sulfate (Na2SO4), the solvent was evaporated. Further, methyl ester product was achieved by distillation under reduced pressure.
Yield: 90%; IR (KBr, cm-1) νmax: 1750(C=O), 682(C-Br). 1H NMR (CDCl3, 400 MHz): δ ppm 7.49 (d, 2H, J = 8.52 Hz, phenyl), 7.34 (d, 2H, J = 8.52 Hz, phenyl), 5.32 (s, 1H, CHBr), 3.78 (s, 1H, ester CH3).
Methyl 2-(4-chlorophenyl)-2-(4-(3-chlorophenyl)-3-oxopiperazin-1-yl)acetate (5)
1-(3-chlorophenyl) piperazin-2-one hydrochloride (5 g, 20.24 mmol) was added to methyl α-bromo (4-chlorophenyl)acetate (5.33 g, 20.24 mmol) in 50 ml of methanol along with sodium bicarbonate (3.4 g, 40.48 mmol) and the mixture was stirred at 80°C. After 6 hours, precipitated solids were separated by filtration, and the solvent was evaporated under reduced pressure. Then, the precipitate was dissolved in ethyl acetate (60 mL) and washed by water (30 mL). The separated organic phase washed with distilled water and cooled to -10°C. A mixture of 10 g of ice and 5 mL concentrated HCl was then added. Finally, the filtered precipitate was dried to obtain the expected product.
Yield: 69%; mp = 100-102°C; IR (KBr, cm-1) νmax: 3045(Aromatic), 1740 (ester C=O), 1675 (amide C=O), 1585 (aromatic C=C), 1290 (C-O). 1H NMR (CDCl3, 500 MHz): δ ppm 7.55-7.33 (m, 8H, phenyl), 4.99 (s, 1H, CHCOOMe), 3.80 (d, 2H, J=18.5 Hz, piperazinone), 3.68 (s, 3H, ester CH3), 3.36 (s, 2H, piperazinone), 3.15 (d, 2H, J=18.5 Hz, piperazinone). 13C NMR (CDCl3, 125 MHz,) δ ppm 169.20, 163.90, 142.81, 134.10, 132.86, 131.04, 130.46, 129.03, 126.61, 125.91, 124.39, 118.21, 68.75, 53.53, 52.64, 47.72, 46.77. MS (ESI): 394.39 [M+H]; Anal.
Calcd. for C19H18Cl2N2O3: C58.04, H 4.62, N 7.11; Found: C 57.91, H 4.62, N 7.15 %.
2-(4-Chlorophenyl)-2-(4-(3-chlorophenyl)-3-oxopiperazin-1-yl)acetic acid (6)
A solution of 5 (5.10g, 13 mmol) and NaOH (1.56 g, 39 mmol) was stirred in 50 mL MeOH/H2O (50:50) overnight at 25°C. After evaporation of the MeOH, the remaining suspension was adjusted with 200mL additional H2O and washed with Et2O (2×100 mL). HCl 1N (pH<3) was added to the residual solution, and extraction was done three times, each of 50 mL of EtOAc. The brine was used for washing the organic layer, and then it was dried by sodium sulfate (Na2SO4). The solvent evaporated under reduced pressure to obtain colorless oil.
Yield: 83%; mp = 219-220°C; IR (KBr, cm-1) νmax: 3300-2300(OH), 1740(Acid C=O), 1675(Amid C=O). 1H NMR (CDCl3, 500 MHz): δppm 7.60-7.33 (m, 8H, Phenyl), 5 (s, 1H, CHCOOH), 3.83 (brs, 2H, piperazinone), 3.35 (s, 2H, piperazinone), 3.27 (brs, 2H, piperazinone).13C NMR (CDCl3, 125 MHz,) δ ppm 169.57, 163.26, 142.68, 134.18, 132.88, 131.23, 130.51, 129.02, 126.73, 125.96, 124.45, 121, 69.06, 53.1, 47.25, 45.19. MS (ESI): 380.09 [M+H]; Anal. Calcd. for C18H16Cl2N2O3: C57.01, H 4.26, N 7.37; Found: C 56.89, H 4.24, N 7.41 %.
General procedure for the synthesis of 7a-7c
2.3 mL of oxalyl chloride (26.5 mmol) was added dropwise to the solution of derivative 6 (5g, 13.19 mmol) in dry dichloromethane (50 mL) and DMF (0.5 mL) at 0oC over 30 minutes. The reaction was stirred 24 hours at 25°C. The dichloromethane was evaporated under vacuum to yield acyl chloride and used without any purification.
0.5 g of acyl halide (1.257 mmol) in dry dichloromethane (3 mL) was added dropwise to a stirred solution of appropriate amine (1.5 mmol) and 0.3 g of triethylamine (3 mmol) in dry dichloromethane (10 mL) at 0°C. The solution was stirred at 25°C for 3.5 hours, and the dichloromethane was evaporated under reduced pressure. The precipitate was dissolved in 10 mL EtOAc and washed with distilled water (3×10 mL). The EtOAc was dried by sodium sulfate (Na2SO4) and filtered. Then, the EtOAc was evaporated under reduced pressure. The purification of obtained products was done using column chromatography eluted with hexane/EtOAc. The yields of reactions ranged between 50 to 80%.
2-(4-Chlorophenyl)-2-[4-(3-chlorophenyl)-3-oxopiperazin-1-yl]-N-hydroxyacetamide (7a)
Yield: 55%; mp = 260°C; IR (KBr, cm-1) νmax: 3300-2500(OH), 3200(NH), 1670(C=O), 1660(C=O). 1H NMR (DMSO-d6, 500 MHz): δ ppm 7.52-7.32 (m, 8H, phenyl), 3.92 (s, 1H, CHCONHOH), 3.48 (dt, 2H, J=33.25 Hz, J=5 Hz, piperazinone), 3.34 (s, 2H, piperazinone), 3.13 (dd, 2H, J=50 Hz, J=16.25 Hz, piperazinone).13C NMR (DMSO-d6, 125 MHz) δ ppm 166.1, 162.17, 143.75, 136.36, 133.28, 133, 128.8, 128.72, 126.7, 126.24, 124.64, 122.54, 69.57, 60.59, 55.8, 49.35. MS (ESI): 395.29 [M+H]; Anal. Calcd. for C18H17Cl2N3O3: C54.84, H 4.36, N 10.65; Found: C 54.66, H 4.34, N 10.69 %.
2-(4-Chlorophenyl)-2-[4-(3-chlorophenyl)-3-oxopiperazin-1-yl]acetamide(7b)
Yield: 79%; mp = 254-256 oC; IR (KBr, cm-1) νmax: 3380-3130(NH2), 1675(C=O), 1645(C=O). 1H NMR (DMSO-d6, 500 MHz): δ ppm 7.38-7.13 (m, 8H, phenyl), 4.54 (s, 1H, CHCONH), 3.66 (t, 2H, J=5.8 Hz, piperazinone), 3.46 (s, 2H, piperazinone), 3.04 (t, 2H, J=5.8 Hz, piperazinone). 13C NMR (DMSO-d6, 125 MHz) δ ppm 171.02, 162.77, 142.57, 136.89, 134.11, 132.76, 131.21, 130.74, 128.54, 123.4, 119.42, 119.11, 69.48, 55.33, 49.03. MS (ESI): 379.25 [M+H]; Anal. Calcd. for C18H17Cl2N3O2: C57.16, H 4.54, N 11.13; Found: C 56.99, H 4.54, N 11.09 %.
2-(4-Chlorophenyl)-2-[4-(3-chlorophenyl)-3-oxopiperazin-1-yl]acetohydrazide(7c)
Yield: 66%; m.p = 256-258 oC; IR (KBr, cm-1) νmax: 3270-3180 (NH2 and NH), 1680(C=O), 1651 (C=O). 1H NMR (DMSO-d6, 500 MHz): δ ppm 7.52-7.31 (m, 8H, phenyl), 3.92 (s, 1H, CHCONHNH2), 3.64 (d, 2H, J=16.35 Hz, piperazinone), 3.42 (s, 2H, piperazinone), 3.12 (d, 1H, J = 16.25 Hz, piperazinone). 13C NMR (DMSO-d6, 125 MHz) δ ppm 168.80, 166.18, 143.74, 136.11, 133.3, 133.09, 131.23, 130.80, 128.77, 126.71, 126.25, 124.62, 70.43, 55.78, 49.33, 47.53. MS (ESI): 394.19 [M+H]; Anal. Calcd. for C18H18Cl2N4O2: C54.97, H 4.62, N 14.26; Found: C 54.79, H 4.61, N 14.21 %.
General procedure for the synthesis of 7d-7g
To stirred, dry, and 15 mL boiling acetonitrile solution of an appropriate amine (3.14 mmol) was added a solution of acyl halide (0.5g, 1.257 mmol) in 5 mL dry acetonitrile dropwise. The solution was refluxed for 3 hours. Then, the acetonitrile was evaporated under reduced pressure. The crude product was dissolved in EtOAc (15 mL) and washed using water (3×10 mL). The EtOAc was dried over sodium sulfate (Na2SO4) and evaporated. Finally, products were purified through recrystallization using EtOAc/n-hexane as the mobile phase.
N-(Aminocarbonyl)-2-(4-chlorophenyl)-2-[4-(3-chlorophenyl)-3-oxopiperazin-1-yl]acetamide (7d)
Yield: 51%; mp = 282-284°C; IR (KBr, cm-1) νmax: 3375-3125(NH2, NH), 1680(C=O), 1651(C=O), 1635(C=O). 1H NMR (DMSO-d6, 500 MHz) δ ppm 7.49-7.30 (m, 8H, phenyl), 4.23 (s, 1H, CHCONH), 3.37 (d, 2H, J=11 Hz, piperazinone), 3.17 (s, 2H, piperazinone), 3.10 (d, 1H, J=11 Hz, piperazinone). 13C NMR (DMSO-d6, 125 MHz) δ ppm 173.13, 172.27, 166.78, 143.57, 138.99, 132.82, 131.18, 130.42, 130.34, 127.65, 126.09, 125.76, 124.13, 75.5, 55.81, 49.23, 47.35. MS (ESI): 421.99 [M+H]; Anal. Calcd. for C19H18Cl2N4O3: C54.17, H 4.30, N 13.30; Found: C 54.36, H 4.31, N 13.27 %.
1-(3-Chlorophenyl)-4-[1-(4-chlorophenyl)-2-(1H-imidazol-1-yl)-2-oxoethyl]piperazin-2-one (7e)
Yield: 35%; mp = 206-209°C; IR (KBr, cm-1) νmax: 1690 (C=O), 1650(C=O). 1H NMR (DMSO-d6, 400 MHz): δ ppm 7.85 (s, 1H, imidazole), 7.79-6.92 (m, 10H, phenyl and imidazole), 4.39 (s, 1H, CHCONH), 3.59 (d, 2H, 2H, J=9.2 Hz, piperazinone), 3.43 (s, 2H, piperazinone), 3.05 (d, 2H, J=9.2 Hz, piperazinone). 13C NMR (DMSO-d6, 75 MHz) δ ppm 165.15, 157.22, 150.41, 141.65, 136.91, 134.69, 134.36, 130.31, 130.24, 127.79, 126, 123.51, 121.11, 117.59, 51.14, 48.93, 48.57.MS (ESI): 430.29 [M+H]; Anal. Calcd. for C21H18Cl2N4O2: C58.75, H 4.24, N 13.06; Found: C 58.58, H 4.24, N 13.09 %.
N-(aminocarbonothioyl)-2-(4-chlorophenyl)-2-[4-(3-chlorophenyl)-3-oxopiperazin-1-yl]acetamide (7f)
Yield: 63%; mp = 265-267°C; IR (KBr, cm-1) νmax: 3400-3250 (NH2, NH), 1680 (C=O), 1640 (C=O), 1480(C=S). 1H NMR (DMSO-d6, 400 MHz): δ ppm 7.54-7.33 (m, 8H, phenyl), 4.74 (s, 1H, CHCONH), 3.70 (brd, 2H, piperazinone), 3.51 (s, 2H, piperazinone), 3.33 (brd, 2H, piperazinone).13C NMR (DMSO-d6, 75 MHz) δ ppm 187.81, 165.72, 164.5, 142.49, 138.48, 135.64, 134.88, 133.84, 131.26, 128.76, 118.8, 117.72, 50.47, 49.16, 48.18, 47.45.MS (ESI): 438.69 [M+H]; Anal. Calcd. for C19H18Cl2N4O2S: C52.19, H 4.16, N 12.82; Found: C 52.11, H 4.14, N 12.83 %.
N-[amino(imino)methyl]-2-(4-chlorophenyl)-2-[4-(3-chlorophenyl)-3-oxopiperazin-1-yl]acetamide (7g)
Yield: 33%; mp = 265-267 oC; IR (KBr, cm-1) νmax: 3400-3180 (NH2, NH), 1680 (C=O), 1640 (C=O), 1480(C=S). 1H NMR (DMSO-d6, 400 MHz): δ ppm 7.41-7.11 (m, 8H, phenyl), 4.14 (s, 1H, CHCONH), 4.02 (s, 2H, piperazinone), 3.71 (t, 2H, J=5.4 Hz, piperazinone), 3.63 (t, 2H, J=5.4 Hz, piperazinone).13C NMR (DMSO-d6, 125 MHz) δ ppm 166.72, 164.5, 139.59, 137.58, 136.64, 132.88, 130.24, 128.86, 128.76, 120.61, 118.52, 117.87, 52.77, 48.16, 47.48, 43.45.MS (ESI): 421.09 [M+H]; Anal. Calcd. for C19H19Cl2N5O2: C54.30, H 4.56, N 16.66; Found: C 54.14, H 4.55, N 16.63 %.
Growth inhibition assay
Compounds 5, 6,and 7a-7g were evaluated forcytotoxic activity against HT29 (human colonic adenocarcinoma cells), A549 (adenocarcinoma human alveolar basal epithelial cells) as cancerous cell lines and one normal lung MRC-5 cells (normal fetal lung fibroblasts) using MTT assay. Seven concentrations (0.5-1000µM) of each compound were prepared. After seeding the cell suspensions (1 × 105 cells/mL) in 96-well plates, they were incubated at 37°C to adhere to the cells. The cells were treated with mentioned synthesized compounds for 72 hours. Culture medium were removed and MTT [3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] (25 μL, 4 mg/mL in PBS) were added to each well. After 3 hours of incubation at 37°C the medium was removed. By adding 100 μL of dimethyl sulfoxide per well and shaking for 15 min at 37°C, the purple formazan crystals were dissolved. The absorbance of wells was read at 570 nm using plate reader (sunrise Tecan, Austria). The results of each experiment which was done in triplicate in MTT assay were mentioned as mean ± SD.19
Statistical analysis
The IC50 values were measured by GraphPad Prism v5.0.4.533 (GraphPad Software, San Diego, CA, USA).
Results and Discussion
In the research, various novel piperazin-2-one derivatives, which contained guanidine, thiourea, hydrazide, and etc., were synthesized, characterized, and assayed against HT29, A549, and MRC-5 cell lines to study the relationship between structure and cytotoxicity.
Intermediate 2 was synthesized in a 50% yield via the reaction of 4-chlorophenylacetic acid with bromine and phosphorus trichloride in benzene. It was then esterified using a mixture of methanol with sulfuric acid as illustrated in scheme 1.20,21
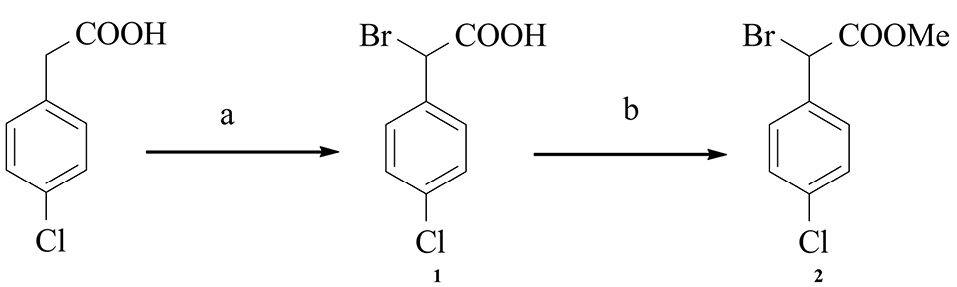
Scheme 1.
a) Br2, PCl3, benzene, reflux, 3 days; b) H2SO4, CH3OH, reflux, Overnight.21
.
a) Br2, PCl3, benzene, reflux, 3 days; b) H2SO4, CH3OH, reflux, Overnight.21
Intermediate 4 and L-778,123 were synthesized according to well-known methods described previously.21,22 scheme 2 displays the synthetic route for intermediate 4.
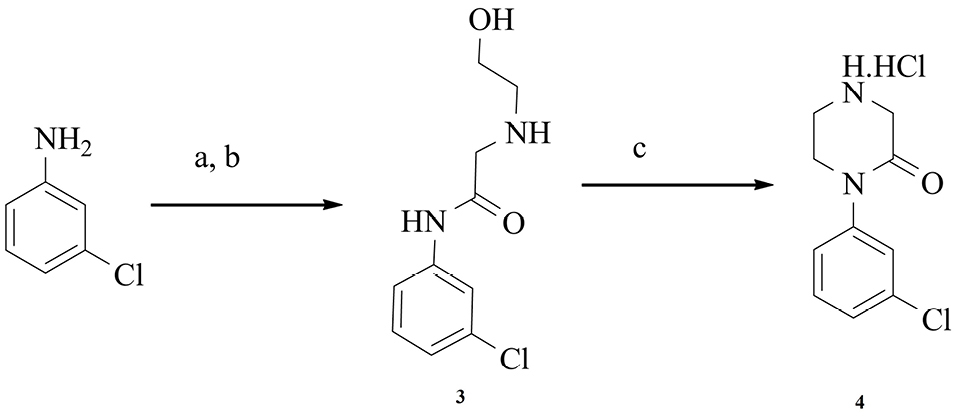
Scheme 2.
a) Chloroacetyl chloride, IpAc 0°C; b) Ethanolamine, IpAc, 55oC, 1 h; c) Tributylphosphine, DIAD, EtAc, -10°C.22,23
.
a) Chloroacetyl chloride, IpAc 0°C; b) Ethanolamine, IpAc, 55oC, 1 h; c) Tributylphosphine, DIAD, EtAc, -10°C.22,23
scheme 3 reveals the synthetic method for the preparation of the title compounds 5, 6, and 7a-g.
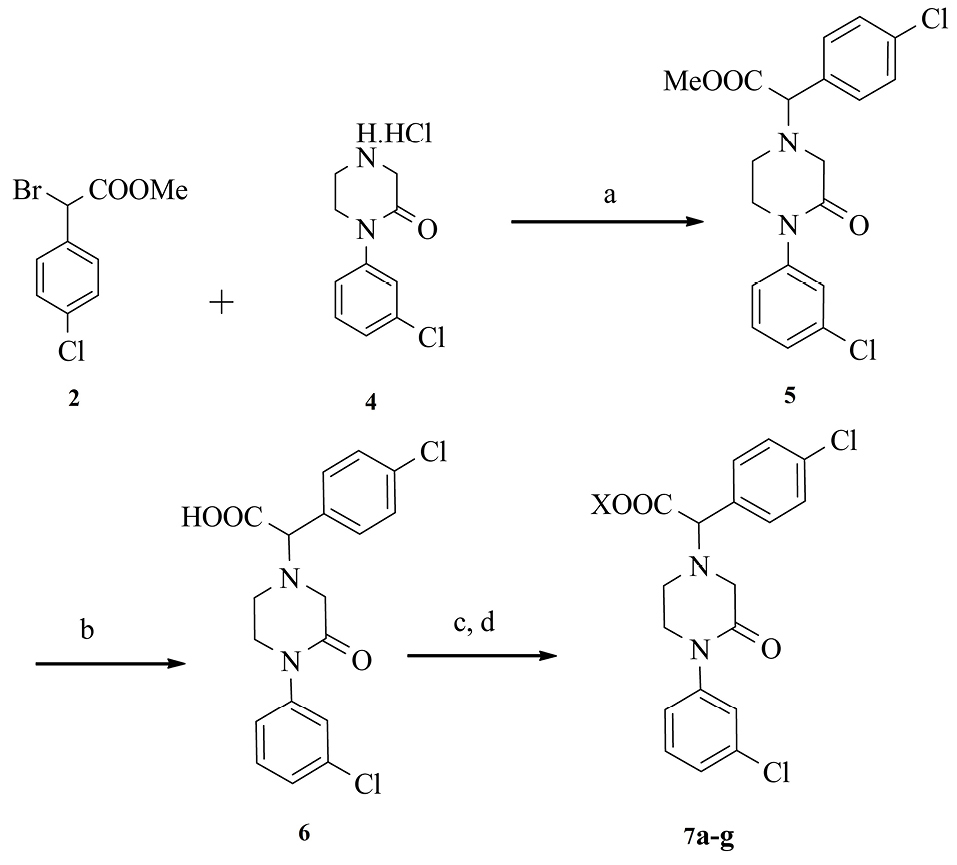
Scheme 3.
a) NaHCO3, MeOH, 80°C, 4 h; b) NaOH, MeOH/H2O, Overnight, r.t; c) Oxalyl chloride, CH2Cl2, DMF, 0°C; d)appropriate amine, CH2Cl2 or CH3CN.
.
a) NaHCO3, MeOH, 80°C, 4 h; b) NaOH, MeOH/H2O, Overnight, r.t; c) Oxalyl chloride, CH2Cl2, DMF, 0°C; d)appropriate amine, CH2Cl2 or CH3CN.
The intermediate 5 was synthesized by the reaction of intermediates 2 and 4 using potassium carbonate (K2CO3) in methanol.24 The derivative 5 was hydrolyzed under basic conditions to 6 derivatives.25 The product 6 was converted to acyl halide by thionyl chloride in dichloromethane and dimethylformamide (DMF).26 The compounds 7a-g were prepared from acyl halide and selected amine in dichloromethane or acetonitrile.27,28
Table 1 reports the results of cytotoxicity as IC50 (µM) of the compounds. The majority of final compounds indicated a significant cytotoxic activity on both cancer cell lines at concentrations <500 µM except for compounds 5 and 6. The guanidine derivative (compound 7g) exhibited the highest cytotoxicity against two investigated cancer cells at concentrations <2 µM, which were lower than doxorubicin on both cell lines. The other potent compounds were the thiourea and hydrazide derivatives (compounds 7fand7c). The cytotoxic activity of compound 7ewith imidazole substituent did not change significantly compared to L-778,123. The compound 7d showed significant cytotoxic effect, but it was less than L-778,123 (as the standard compound). The compounds 7aand7b indicated cytotoxicity near 500 µM. The compounds 5 and 6, however, did not indicate the significant cytotoxic effect on both cells (>1 μM).
Table 1.
Cytotoxic activities (IC50, µM) of intermediate 5, 6 and compounds 7a-7g on A549 (lung cancer), HT-29 (colon cancer), and MRC-5 (normal fetal lung fibroblasts) cells
|
|
|
Compounds
|
X
|
A549
|
HT29
|
MRC-5
|
|
5
|
-OMe |
>1000 |
>1000 |
>1000 |
|
6
|
-OH |
>1000 |
>1000 |
>1000 |
|
7a
|
-NHOH |
431.67±2.85 |
498.17±3.73 |
>1000 |
|
7b
|
-NH2 |
317.44±2.92 |
398.18±1.47 |
>1000 |
|
7c
|
-NHNH2 |
22.67±0.83 |
49.28±0.68 |
125.71±1.26 |
|
7d
|
-NHCONH2 |
195.18±0.93 |
193.74±1.02 |
251.86±1.93 |
|
7e
|
Imidazole |
110±2.51 |
128.99±2.86 |
194.23±1.44 |
|
7f
|
-NHCSNH2 |
5.11±1.41 |
6.21±1.21 |
8.75±0.53 |
|
7g
|
-NHCNHNH2 |
1.3±0.22 |
1.8±0.13 |
5.28±0.41 |
|
L-778,123
|
|
101±2 |
125±2 |
150.70±1.72 |
|
Doxorubicin
|
|
4.1±0.1 |
3±0.1 |
0.85±0.12 |
Compounds 5, 6, 7a, and 7b did not show cytotoxicity against MRC-5. Compounds7f and 7g showed significant cytotoxicity against normal MRC-5 cells. All compounds were significantly more cytotoxic toward both cancer cell lines as compared to normal MRC-5 cells.
The cytotoxic mechanism of these derivatives is not clear. Although it has been suggested that they exert their effects through inhibiting FTase, most synthesized derivatives which had not indicated acceptable cytotoxicity have been effective FTIs and vice versa.13-16,18
The benzyl group of the derivatives can be substituted by different electron-withdrawing groups like nitrile- or chloro- moieties at the para position. Several FTIs such as tipifarnib have chloro group instead of cyano group at this position. These FTIs have indicated higher cytotoxicity than L-778,123. We predicted that the substitution of 4-CN for 4-Cl could improve the cytotoxic activity of the compounds. So, 4-cyanobenzyl was substituted for 4-chlorobenzyl in these derivatives.17,22,23,26,29-31
Lung and colon cancers were selected for cytotoxic activity assessment because they are the most important kinds of cancers causing death over the world according to WHO reports. Besides, HT-29 (colon cancer) and A549 (lung cancer) cells are routine cells in cytotoxicity evaluation in many studies. Also, the cell lines showed wild-type K-ras.32,33
The terminal aliphatic nitrogen atoms of bioisosteres may have an essential effect in cytotoxicity. The cytotoxicity of compound 7e showed that only bioisosteric replacement significantly changes the cytotoxic effect. Replacement of imidazole with guanidine, thiourea, and hydrazide (compounds 7g, 7f, and 7c) led to significantly better cytotoxic effect on all three cell lines in comparison with L-778,123. The improvement in the cytotoxic effect may be as a result of the fact that terminal atoms have higher electron density.34-36 It has been demonstrated that bioisosteres of imidazole show cytotoxicity through different mechanisms like inhibition of inosine monophosphate dehydrogenase by guanidine-based compounds. The cytotoxic effect of potent compounds (7c, 7f, and 7g) can be related only to the cytotoxicity of imidazole bioisosteres.16,37-39 Other substitutes did not significantly increase the cytotoxic effect in comparison with the above mentioned potent compounds. It can be attributed to the amide groups with lower electron density. Compound 7b indicated better cytotoxic activity than compound 7a. It seems that the substitution of hydroxyl on the amine group can decrease the electron density on terminal nitrogen. Finally, it seems rearrangement of groups based on tipifarnib structure can increase the cytotoxicity against three cell lines. The IC50 of the guanidine derivative in this study showed significant decrease against all cell lines in comparison with previous work.25
Nevertheless, for understanding the precise mechanisms of the action of the synthesized compounds, further studies are needed.
Conclusion
A set of bioisosteres of imidazole containing-derivatives 1-(3-chlorophenyl)piperazin-2-one group were prepared, characterized by various methods including IR, 1HNMR, 13CNMR, and Mass spectroscopy, and tested for cytotoxic activity. The piperazinone derivatives with guanidine substituent (7g) showed the highest potency against all cell lines. Notably, it showed better cytotoxicity than doxorubicin against both cancer cell lines. So it can be a proper lead compound to design novel anticancer compounds. The electron density of terminal atoms of bioisosteres may be an essential factor in the cytotoxic activity. Finally, rearrangement of groups similar to tipifarnib structure can significantly enhance the cytotoxicity. All synthetic compounds showed lower cytotoxicity against healthy cell lines than cancer cell lines.
Ethical Issues
Not applicable.
Conflict of Interest
The authors declare no conflict of interest in this article’s content.
Acknowledgments
The faculty of pharmacy and the tuberculosis and lung disease research center, Tabriz University of medical sciences, have supported this work financially.
References
- Aliabadi A, Shamsa F, Ostad SN, Emami S, Shafiee A, Davoodi J. Synthesis and biological evaluation of 2-phenylthiazole-4-carboxamide derivatives as anticancer agents. Eur J Med Chem 2010; 45(11):5384-9. doi: 10.1016/j.ejmech.2010.08.063 [Crossref] [ Google Scholar]
- Ghanbarzadeh S, Ghasemi S, Shayanfar A, Ebrahimi-Najafabadi H. 2D-QSAR study of some 2,5-diaminobenzophenone farnesyltransferase inhibitors by different chemometric methods. EXCLI J 2015; 14:484-95. doi: 10.17179/excli2015-177 [Crossref] [ Google Scholar]
- Bolchi C, Pallavicini M, Rusconi C, Diomede L, Ferri N, Corsini A. Peptidomimetic inhibitors of farnesyltransferase with high in vitro activity and significant cellular potency. Bioorg Med Chem Lett 2007; 17(22):6192-6. doi: 10.1016/j.bmcl.2007.09.015 [Crossref] [ Google Scholar]
- Vaidya M, Weigt M, Wiese M. 3D-QSAR with the aid of pharmacophore search and docking-based alignments for farnesyltransferase inhibitors. Eur J Med Chem 2009; 44(10):4070-82. doi: 10.1016/j.ejmech.2009.04.045 [Crossref] [ Google Scholar]
- Appels NM, Beijnen JH, Schellens JH. Development of farnesyl transferase inhibitors: a review. Oncologist 2005; 10(8):565-78. doi: 10.1634/theoncologist.10-8-565 [Crossref] [ Google Scholar]
- Angibaud P, Mevellec L, Meyer C, Bourdrez X, Lezouret P, Pilatte I. Impact on farnesyltransferase inhibition of 4-chlorophenyl moiety replacement in the Zarnestra series. Eur J Med Chem 2007; 42(5):702-14. doi: 10.1016/j.ejmech.2006.12.007 [Crossref] [ Google Scholar]
- Puntambekar DS, Giridhar R, Yadav MR. Insights into the structural requirements of farnesyltransferase inhibitors as potential anti-tumor agents based on 3D-QSAR CoMFA and CoMSIA models. Eur J Med Chem 2008; 43(1):142-54. doi: 10.1016/j.ejmech.2007.02.003 [Crossref] [ Google Scholar]
- Equbal T, Silakari O, Rambabu G, Ravikumar M. Pharmacophore mapping of diverse classes of farnesyltransferase inhibitors. Bioorg Med Chem Lett 2007; 17(6):1594-600. doi: 10.1016/j.bmcl.2006.12.087 [Crossref] [ Google Scholar]
- Shayanfar A, Ghasemi S, Soltani S, Asadpour-Zeynali K, Doerksen RJ, Jouyban A. Quantitative structure-activity relationships of imidazole-containing farnesyltransferase inhibitors using different chemometric methods. Med Chem 2013; 9(3):434-48. doi: 10.2174/1573406411309030014 [Crossref] [ Google Scholar]
- Huang CY, Stauffer TM, Strickland CL, Reader JC, Huang H, Li G. Guiding farnesyltransferase inhibitors from an ECLiPS library to the catalytic zinc. Bioorg Med Chem Lett 2006; 16(3):507-11. doi: 10.1016/j.bmcl.2005.10.070 [Crossref] [ Google Scholar]
- Hasvold LA, Wang W, Gwaltney SL 2nd, Rockway TW, Nelson LT, Mantei RA. Pyridone-containing farnesyltransferase inhibitors: synthesis and biological evaluation. Bioorg Med Chem Lett 2003; 13(22):4001-5. doi: 10.1016/j.bmcl.2003.08.058 [Crossref] [ Google Scholar]
- Gilleron P, Millet R, Houssin R, Wlodarczyk N, Farce A, Lemoine A. Solid-phase synthesis and pharmacological evaluation of a library of peptidomimetics as potential farnesyltransferase inhibitors: an approach to new lead compounds. Eur J Med Chem 2006; 41(6):745-55. doi: 10.1016/j.ejmech.2006.03.017 [Crossref] [ Google Scholar]
- Bolchi C, Pallavicini M, Bernini SK, Chiodini G, Corsini A, Ferri N. Thiazole- and imidazole-containing peptidomimetic inhibitors of protein farnesyltransferase. Bioorg Med Chem Lett 2011; 21(18):5408-12. doi: 10.1016/j.bmcl.2011.07.003 [Crossref] [ Google Scholar]
- Maiti KK, Jeon OY, Lee WS, Chung SK. Design, synthesis, and delivery properties of novel guanidine-containing molecular transporters built on dimeric inositol scaffolds. Chemistry 2007; 13(3):762-75. doi: 10.1002/chem.200600898 [Crossref] [ Google Scholar]
- Zhang HZ, Crogan-Grundy C, May C, Drewe J, Tseng B, Cai SX. Discovery and structure-activity relationships of (2-(arylthio)benzylideneamino)guanidines as a novel series of potent apoptosis inducers. Bioorg Med Chem 2009; 17(7):2852-8. doi: 10.1016/j.bmc.2009.02.029 [Crossref] [ Google Scholar]
- Iwanowicz EJ, Watterson SH, Liu C, Gu HH, Mitt T, Leftheris K. Novel guanidine-based inhibitors of inosine monophosphate dehydrogenase. Bioorg Med Chem Lett 2002; 12(20):2931-4. doi: 10.1016/s0960-894x(02)00600-5 [Crossref] [ Google Scholar]
- Popp FD, Swarz H. Synthesis of potential anticancer agents VI urea and thiourea mustards1,2. J Org Chem 1961; 26(11):4764-5. doi: 10.1021/jo01069a562 [Crossref] [ Google Scholar]
- Omar AM, Farghaly AM, Hazzai AA, Eshba NH, Sharabi FM, Daabees TT. Thiourea and thiosemicarbazide derivatives structurally related to hexestrol: synthesis and anticancer and other pharmacological properties. J Pharm Sci 1981; 70(9):1075-9. doi: 10.1002/jps.2600700928 [Crossref] [ Google Scholar]
- Ghasemi S, Davaran S, Sharifi S, Asgari D, Abdollahi A, Shahbazi Mojarrad J. Comparison of cytotoxic activity of L778123 as a farnesyltranferase inhibitor and doxorubicin against A549 and HT-29 cell lines. Adv Pharm Bull 2013; 3(1):73-7. doi: 10.5681/apb.2013.012 [Crossref] [ Google Scholar]
-
Bouisset M, Radisson J. Process for the preparation of α-bromo-phenylacetic acids. US5036156, 1991.
-
Carpino LA, McAdams LV III. α,α′‐Dibromodibenzyl Sulfone. In: Organic Synthesis. Vol 50. Wiley; 2003.50. 10.1002/0471264180.os050.11
- Maligres PE, Waters MS, Weissman SA, McWilliams JC, Lewis S, Cowen J. Preparation of a clinically investigated ras farnesyl transferase inhibitor. J Heterocycl Chem 2003; 40(2):229-41. doi: 10.1002/jhet.5570400206 [Crossref] [ Google Scholar]
-
Askin D, Lewis S, Weissman SA. Process for the synthesis of substituted piperazinones via Mitsunobu reaction. US6160118, 2000.
- Shahbazi Mojarrad J, Zamani Z, Nazemiyeh H, Ghasemi S, Asgari D. Synthesis of novel 1, 4-dihydropyridine derivatives bearing biphenyl-2’-tetrazole substitution as potential dual angiotensin II receptors and calcium channel blockers. Adv Pharm Bull 2011; 1(1):1-9. doi: 10.5681/apb.2011.001 [Crossref] [ Google Scholar]
- Ghasemi S, Sharifi S, Davaran S, Danafar H, Asgari D, Mojarrad JS. Synthesis and cytotoxicity evaluation of some novel 1-(3-chlorophenyl) piperazin-2-one derivatives bearing imidazole bioisosteres. Aust J Chem 2013; 66(6):655-60. doi: 10.1071/CH13031 [Crossref] [ Google Scholar]
- Simoneau B, Lavallée P, Anderson PC, Bailey M, Bantle G, Berthiaume S. Discovery of non-peptidic P2-P3 butanediamide renin inhibitors with high oral efficacy. Bioorg Med Chem 1999; 7(3):489-508. doi: 10.1016/s0968-0896(98)00265-x [Crossref] [ Google Scholar]
- Pan C, Zeng X, Guan Y, Jiang X, Li L, Zhang H. Design and synthesis of brazilin-like compounds. Synlett 2011; 2011(03):425-9. doi: 10.1055/s-0030-1259511 [Crossref] [ Google Scholar]
- Zhang Y, Williams W, Torrence-Campbell C, Bowen WD, Rice KC. Characterization of novel N,N’-disubstituted piperazines as sigma receptor ligands. J Med Chem 1998; 41(25):4950-7. doi: 10.1021/jm980143k [Crossref] [ Google Scholar]
- Grzyb JA, Shen M, Yoshina-Ishii C, Chi W, Brown RS, Batey RA. Carbamoylimidazolium and thiocarbamoylimidazolium salts: novel reagents for the synthesis of ureas, thioureas, carbamates, thiocarbamates and amides. Tetrahedron 2005; 61(30):7153-75. doi: 10.1016/j.tet.2005.05.056 [Crossref] [ Google Scholar]
- Grethe G, Lee HL, Uskokovic M, Brossi A. Syntheses in the isoquinoline series Synthesis of 2,3-dihydro-4(1H)-isoquinolones. J Org Chem 1968; 33(2):491-4. doi: 10.1021/jo01266a003 [Crossref] [ Google Scholar]
- Shendage DM, Fröhlich R, Haufe G. Highly efficient stereoconservative amidation and deamidation of alpha-amino acids. Org Lett 2004; 6(21):3675-8. doi: 10.1021/ol048771l [Crossref] [ Google Scholar]
- Li Q, Li T, Woods KW, Gu WZ, Cohen J, Stoll VS. Benzimidazolones and indoles as non-thiol farnesyltransferase inhibitors based on tipifarnib scaffold: synthesis and activity. Bioorg Med Chem Lett 2005; 15(11):2918-22. doi: 10.1016/j.bmcl.2005.03.049 [Crossref] [ Google Scholar]
- Pingaew R, Tongraung P, Worachartcheewan A, Nantasenamat C, Prachayasittikul S, Ruchirawat S. Cytotoxicity and QSAR study of (thio)ureas derived from phenylalkylamines and pyridylalkylamines. Med Chem Res 2013; 22(8):4016-29. doi: 10.1007/s00044-012-0402-6 [Crossref] [ Google Scholar]
- Angibaud P, Saha AK, Bourdrez X, End DW, Freyne E, Lezouret P. 4-methyl-1,2,4-triazol-3-yl heterocycle as an alternative to the 1-methylimidazol-5-yl moiety in the farnesyltransferase inhibitor ZARNESTRA. Bioorg Med Chem Lett 2003; 13(24):4361-4. doi: 10.1016/j.bmcl.2003.09.043 [Crossref] [ Google Scholar]
- Angibaud P, Bourdrez X, Devine A, End DW, Freyne E, Ligny Y. 5-imidazolyl-quinolinones, -quinazolinones and -benzo-azepinones as farnesyltransferase inhibitors. Bioorg Med Chem Lett 2003; 13(9):1543-7. doi: 10.1016/s0960-894x(03)00180-x [Crossref] [ Google Scholar]
- Nguyen DN, Stump CA, Walsh ES, Fernandes C, Davide JP, Ellis-Hutchings M. Potent inhibitors of farnesyltransferase and geranylgeranyltransferase-I. Bioorg Med Chem Lett 2002; 12(9):1269-73. doi: 10.1016/s0960-894x(02)00154-3 [Crossref] [ Google Scholar]
- Kedzia BB, Armendarez PX, Nakamoto K. Infra-red spectra and normal co-ordinate analyses of metal biuret complexes. Journal of Inorganic and Nuclear Chemistry 1968; 30(3):849-60. doi: 10.1016/0022-1902(68)80446-4 [Crossref] [ Google Scholar]
- Okudela K, Hayashi H, Ito T, Yazawa T, Suzuki T, Nakane Y. K-ras gene mutation enhances motility of immortalized airway cells and lung adenocarcinoma cells via Akt activation: possible contribution to non-invasive expansion of lung adenocarcinoma. Am J Pathol 2004; 164(1):91-100. doi: 10.1016/s0002-9440(10)63100-8 [Crossref] [ Google Scholar]
- Brink M, de Goeij AF, Weijenberg MP, Roemen GM, Lentjes MH, Pachen MM. K-ras oncogene mutations in sporadic colorectal cancer in The Netherlands Cohort Study. Carcinogenesis 2003; 24(4):703-10. doi: 10.1093/carcin/bgg009 [Crossref] [ Google Scholar]