Advanced pharmaceutical bulletin. 13(1):176-187.
doi: 10.34172/apb.2023.019
Research Article
Ferroptosis as a Potential Cell Death Mechanism Against Cisplatin-Resistant Lung Cancer Cell Line
Morteza Golbashirzadeh 1, 2  , Hamid Reza Heidari 1, 2, * , Mehdi Talebi 3, Ahmad Yari Khosroushahi 1, 4, *
, Hamid Reza Heidari 1, 2, * , Mehdi Talebi 3, Ahmad Yari Khosroushahi 1, 4, *
Author information:
1Drug Applied Research Center, Tabriz University of Medical Sciences, Tabriz, Iran.
2Department of Pharmaceutical Biotechnology, Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran.
3Hematology and Oncology Research Center, Department of Applied Cell Sciences, School of Advanced Medical Sciences, Tabriz University of Medical Sciences, Tabriz, Iran.
4Department of Medical Nanotechnology, Faculty of Advanced Medical Science, Tabriz University of Medical Sciences, Tabriz, Iran.
Abstract
Purpose:
Drug resistance is a challenging issue in cancer chemotherapy. Cell death induction is one of the main strategies to overcome chemotherapy resistance. Notably, ferroptosis has been considered a critical cell death mechanism in recent years. Accordingly, in this study, the different cell death strategies focused on ferroptosis have been utilized to overcome cisplatin resistance in an in vitro lung cancer model.
Methods: The physiological functions of Akt1 and GPX4, as critical targets for ferroptosis and apoptosis induction, were suppressed by siRNA or antagonistic agents in resistant A549 cells. Afterward, the interventions’ impacts on cell viability and reactive oxygen species (ROS) amount were analyzed by flow cytometry. Moreover, the alteration in the relevant gene and protein expression levels were quantified using Real-time PCR and western blot methods.
Results: The result showed that the treatment with Akt1 siRNA reversed the cisplatin resistance in the A549 cell line through the induction of apoptosis. Likewise, the combination treatment of the GPX4 siRNA or FIN56 as ferroptosis inducers alongside cisplatin elevated ROS’s cellular level, reduced the cellular antioxidant genes level and increased the cisplatin cytotoxic effect.
Conclusion: In conclusion, our study indicated that ferroptosis induction can be considered a promising cell death strategy in cisplatin-resistant cancer cells.
Keywords: Drug resistance, Cisplatin, Apoptosis, Ferroptosis, Gene silencing
Copyright and License Information
©2023 The Authors.
This is an Open Access article distributed under the terms of the Creative Commons Attribution (CC BY), which permits unrestricted use, distribution, and reproduction in any medium, as long as the original authors and source are cited. No permission is required from the authors or the publishers.
Introduction
Lung cancer is one of the fatal types of cancers worldwide.1 Non-small cell lung carcinoma (NSCLC) is the most common subtype of lung cancer with a high prevalence in the clinic.1,2 Based on Medscape (https://www.medscape.com), only 30% of NSCLC tumors have localized properties. Chemotherapy, instead of surgery/radiation therapy, is considered the first step of the treatment approach. Cisplatin is usually included as a first-line medicine in most NCLC chemotherapy protocols.3-5 Cisplatin exerts its effects by forming crosslinks in the DNA and inhibiting the DNA replication, G2/M phase cell cycle arrest, and inducing apoptosis.6,7 Nowadays, chemoresistance is the main reason for Cisplatin treatment failure.8,9 Galluzzi et al10 have classified cisplatin resistance mechanisms as pre-target resistance (e.g., drug efflux), on-target resistance (e.g., enhanced DNA repair machinery), post-target resistance (e.g., alternation in the drugs’ mechanism of action), and off-target resistance (alterations in compensatory signaling pathways); which were summarized in Table 1.
Table 1.
Mechanism of Cisplatin resistance
|
Cisplatin resistance
|
Mechanism of action
|
Reference
|
| Pre-target resistance |
Avoiding to create cisplatin DNA adduct by decreasing cellular accumulation and efflux the cisplatin outside of the cell |
7,10,11
|
| On-target resistance |
Enhanced DNA repair machinery and increase toleration of DNA |
| Post-target resistance |
Alternation or signaling pathway after DNA deficiency through cisplatin exposure |
| Off-target resistance |
An indirect cellular mechanism side effect that is not directly relative to cisplatin exposure but causes evasion of Apoptosis and Cisplatin cell death |
Among various cellular signaling pathways, the PI3K-AKT has a higher impact on cancer progression and drug resistance. The PI3K-AKT regulates survival, differentiation, proliferation, migration, and chemoresistance in cancer cells. Consequently, targeting PI3K-AKT pathways has been considered a good strategy for battling cancer through Apoptosis induction.12,13
However, as several other molecular pathways contribute to cancer survival and chemoresistance, overcoming the drug resistance for Apoptosis induction in different types of cancers might be challenging.8,9,14 So, opting for another cell death strategy might help re-sensitize these immortal cells.15,16
Numerous types of cell death strategies have been implemented in cancer therapy research.16-19 Previous studies have remarkably revealed that reactive oxygen species (ROS) over-accumulation is a predominant phenomenon in cancer cells.17-21 Therefore, ROS-dependent, Caspase-independent programmed cell death, “ferroptosis,” attracted the researcher’s attention.22,23
Particular criteria such as iron accumulation, lipid peroxidation, loss of mitochondrial function, and membrane integrity are ferroptosis hallmarks.23 Several compounds and siRNAs induce ferroptosis by interfering in the role of glutathione-peroxidase 4 (GPX4), Mevalonate pathway, cysteine-glutamate-anti-porters, and mitochondrial transporters.24-26 In this regard, FIN56, as one of the famous ferroptosis inducers, interacts with the squalene synthase enzyme in the Mevalonate-pathway, which leads to cytoplasmic CoQ10 depletion.27,28 Moreover, FIN56 indirectly promotes GPX4 degradation and causes cellular oxidants accumulation.29,30 The cellular CoQ10 depletion, besides higher amounts of cytoplasmic ROS, results in the peroxidation of cellular phospholipids, accumulation of the lipid ROS, and finally, ferroptosis induction.31
Interestingly, ferroptosis induction by approved clinical anticancer drugs such as sulfasalazine, sorafenib, lapatinib, temozolomide, cisplatin,21 and even by cytotoxic T-cells26 have been reported.
Considerable researches indicate that ferroptosis inducers would attain a good percentage of the novel cancer therapeutics,32,33 especially in chemotherapy resistance forms. Favorably, Roh et al have reported that ferroptosis induction through silencing specific genes such as cystine-glutamate-antiporter (xCT) can increase cisplatin’s efficacy in cisplatin-resistant cancer cells.16 Similarly, according to Sugiyama, xCT inhibitor sulfasalazine eradicates paclitaxel-resistant uterine serous carcinoma.34 Likewise, GPX4 siRNA was used for ferroptosis induction in chemoresistance aggressive Panc-1 cancer stem-like cells.26
Despite these achievements, ferroptosis’s effectiveness vs. apoptosis in eradicating cancer resistance cells was not addressed. Therefore, this study tested the efficacy of these two cell death strategies combined with Cisplatin drug in the cisplatin-resistant A549 as the NSCLC model. At first, ferroptosis effectiveness was examined using GPX4 siRNA and FIN56 agents; then, apoptosis efficacy was investigated with AKT1 siRNA. Finally, the ability of these coadministrations in eradicating resistance A549 cells was compared.
Materials and Methods
Cell culture
The cisplatin-resistant lung cancer cell line (A549 CDDP) was generously gifted by Dr. Roya Salehi, faculty of Advanced Medical Science, Tabriz University of Medical Science, Tabriz, Iran. The normal human foreskin fibroblasts (HFFs) cell line was purchased from the National Cell Bank Pasteur institute of Iran. The cell lines were cultured in RPMI-1640 medium (Gibco, MD, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco, MD, USA) and (penicillin 100 U/mL and streptomycin 100 µg/mL) (Inoclon Co, Iran, 12PS2-100) at 37°C, humidified 5% CO2.
Cell death induction and Cytotoxicity assay
To assess the possibility of using the ferroptosis strategy in combating cancerous resistance cells, we applied both chemical (FIN56) and biological (GPX4 siRNA) treatments. Moreover, Akt1 siRNA was used to evaluate the effect of the Apoptosis induction strategy in this battle.
MTT test was performed to determine the effective dose of FIN56 against the resistant A549 and HFF normal cell lines. In brief, the resistant A549 and HFF cell lines were seeded with the cell density of 1 × 104 cells/well in 96-well microplates and were treated with different FIN56 concentrations (0, 5, 10, 12, 14, 18, 20, 22, 25, 30, 35 μM) for 48 hours, and subsequently subjected to the MTT assay as previously described.35
The GPX4 and Akt1 siRNAs (Table 2) were designed by the siRNA direct website (http://design.RNAi.jp/); and purchased from Eurofins Genomics Company (Ebersberg, Germany). The siRNA transfection procedure was conducted as in the previous study.36 In brief: the resistant A549 cell line was seeded in a 6-well plate one day before transfection at an initial density of 0.7 × 105 cells/well. Then, based on the numerous previous published papers,37-39 100 nM of each siRNAs were complexed with HiPerFect reagent (Qiagen, Germany) in a serum-free media, and mixtures were applied to the cells. After one hour of incubation, treatment media was removed, and the cells were washed with PBS and finally further incubated in the complete media for 48 hours.40
Table 2.
The sequence of siRNA
|
Gene
|
siRNA sequence
|
Length
|
| AKT-1 |
S: 5′- CCAUGAACGAGUUUGAGUACC -3′ |
21 nt |
| A: 5′- UACUCAAACUCGUUCAUGGUC -3′ |
| GPX4 |
S: 5′- CUACAACGUCAAAUUCGAUAU -3′ |
21 nt |
| A: 5′- AUCGAAUUUGACGUUGUAGCC -3′ |
Ferroptosis assay
Cellular lipid ROS and total ROS were assessed after treating cells with FIN56 and GPX4 siRNA by DCFDA and Boron dipyrromethene (BODIPY) dyes according to published protocols41; The emitted fluorescence was detected by a FACSCalibur flow-cytometry (Becton Dickinson, USA).
Apoptosis assay
Determination and analysis of Apoptosis after treatment by Akt1 siRNA and cisplatin has been performed by Annexin V FITC/PI test flowed by published protocol.42
Western blot analysis
The Akt1 and GPX4 proteins’ expression was measured by western blot analysis after siRNA transfection followed by the published protocol.43 In a few words, proteins were lysed for 10 minutes on ice after extraction. Then, the extracted proteins were separated by 12.5% SDS-PAGE and transferred to a polyvinylidene difluoride membrane blocked by BSA. The blocked membrane was incubated with the desired primary antibodies. Finally, horseradish peroxidase-conjugated secondary antibodies were applied with ECL reagent to the reaction based on the manufacturer’s instructions.
RNA isolation, cDNA synthesis, and real-time PCR
Real-time PCR was performed for determining the expression of AKT1, GPX4, Nerf2, and CoQ10 after treatment by FIN56 and the siRNAs in relation to GAPDH as the internal control. Resistance A549 cell line was seeded in a 6-well plate and treated as mentioned in the previous section. Total cellular RNA isolation and cDNA synthesis were performed based on our previously published paper.36 According to the manufacturer’s instructions, the total RNA was extracted by Triazole (GeneAll Biotech, South Korea) reagent. The amount and purity of total RNA were measured by Nanodrop 260/280 nm (Thermo ScientificTM NanoDrop). To synthesize cDNA, 1 µg of total mRNA was used based on a commercially available protocol of BioFact cDNA synthesis kit (Daejeon, South Korea).44 Amplification and alternation of target genes were performed by StepOneTM Real-Time PCR System instrument (Applied Biosystems, USA) with SYBR Green detection system. Suitable primers were designed by NCBI primer blast (https://www.ncbi.nlm.nih.gov/tools/primer-blast), as mentioned in Table 3. The amplification reaction was performed in a 20 μL final volume which contained 1 μL cDNA sample, 2 μL F and R primers (20 pmol), 10 μL Master-mix, and 7 μL RNAs free water. The PCR program was carried out for 40 cycles: first denaturation time at 95°C for 20 minutes, which is followed by 40 cycles of 95°C for 20 seconds, an ideal annealing temperature (Table 3) for 30 seconds; and 72°C for 10 seconds. Afterward, to acquire melting curves, the temperature increased step by step from 65°C to 95°C. Finally, the relative expression of genes was calculated using the ∆∆ Ct method.
Table 3.
List of primers used for detecting specific RNAs using real-time PCR
|
Gene
|
Sequence
|
Annealing
|
Fragment length
|
|
GAPDH
|
F: 5′-TTGACCTCAACTACAGGTTTACA -3′
R: 5′-GCTCCTCCTGGAAGATGGTGATG -3′ |
60°C |
100 |
| GPX4 |
F: 5′-TGGGAAATGCCATCAAGTGGA-3′
R: 5′-GGGCAGGTCCTTCTCTATCAC-3′ |
60°C |
123 |
|
AKT1
|
F: 5′-GAGTTCACGGCCCAGATGAT-3′
R: 5′-CGAGTAGGAGAACTGGGGGA-3′ |
57°C |
105 |
|
Nrf2
|
F: 5′-ATGCCACAGGACATTGAGCA-3′
R: 5′-TTGGCTTCTGGACTTGGAAC-3′ |
60°C |
119 |
|
CoQ10B
|
F: 5′-TTGGATTTCCACCTGTGTTG-3′
R: 5′-CGCCAAATAGTCTCCAAATGA-3′ |
59°C |
118 |
F: forward primer, R: reverse primer.
Trypan blue exclusion and Cell viability assay
The Trypan blue exclusion assay was conducted to evaluate the viability of cell lines after treatments. The resistant A549 cell line was seeded in a 6-well plate. Cells separately were treated with FIN56 (5μM), the Akt1 siRNA (100 nM), or the GPX4 siRNA (100 nM), along with cisplatin (1μM) for 48 hours to set up a combination effect. Then, cells were trypsinized and incubated with Trypan Blue solution (0.4% Trypan Blue, Merck, Germany) for 10 minutes. The percentage of viable and dead cells was measured by FACSCalibur flow-cytometry (Becton Dickinson, USA).
Results and Discussion
Ferroptosis and apoptosis against chemotherapy-resistance lung cancer cells
Several genetically or epigenetically alterations in cancer cells, like gene rearrangements, pathogenic gene mutations, gene expression, post-transcriptional and translational regulation by non-coding RNAs, are responsible for the heterogeneity of different cancers.45-52 These heterogeneities make it impossible to provide a unique magic bullet for cancer treatment. Meanwhile, due to cancer cells’ ever-changing nature, the inherited or acquired resistance forms of cancer cells accumulated in the tumor cells’ environment, which led to a relapse of resistant tumors.53,54
Considering the clinical data of NSCLC in the NCBI ClinVar database, the primary oncogenic driver activating mutations frequently occur in EGFR, HER2, MET, BRAF, RAS, PIK3CA, and MAP2K1 genes. Moreover, chromosomal instabilities, translocation, and fusion of oncogenes such asanaplastic lymphoma kinase (ALK) greatly impact cancer cell generation.13,54,55
Several genetic expression alterations and mutations establish inherited or acquired resistance NSCLC by altering cancer-related proteins’ activity or therapeutic target site (on target resistance). For example, ERCC1 and RRM1 repairing proteins’ overexpression has been associated with gemcitabine and cisplatin resistance tumors. The mutated EGFR, BRAF, ALK, KRAS cancer cells are resistant to various tyrosine kinase inhibitors (TKIs)56-59; and, losing the transmembrane domain of PD-L1 in some mRNA splicing variants led to resistance to anti-PD-1 treatment.60
Moreover, overexpression of the EGFR, c-MET, HER2, FGFR3, and AXL TKIs serves as compensatory signaling pathways such as PI3K-Akt, RAS-ERK, STAT establish off-target resistance to the TKI’s in NSCLC patients.61-65
Additionally, the role of long and small non-coding RNAs in NSCLC chemotherapy resistance is undeniable. Mainly LncRNAs, through alternating the expression of drug efflux proteins, apoptosis, and autophagy modulating proteins, can establish the on-target resistance in NSCLC tumors.66 Besides the roles mentioned above, LncRNAs by induction of cancer stem-cell-like phenotypes and Epithelial-mesenchymal transition can augment the compensatory signaling pathways and establish the off-target resistance in NSCLC.67
The A549 cell line, isolated from human lung alveolar epithelial cell carcinoma, is considered one of the standard NSCLC models for in vitro chemotherapy studies.68 Different cancer cell sub-populations with various genetic mutations, phenotypes, and sensitivity to chemotherapeutic drugs are present in this heterogeneous lung cancer model.69-71
The clinically relevant cisplatin concentration is about 14 µM in the patient’s plasma,72 which is remarkably lower than the expected IC50 of 252.7 µM in the A549 resistance cell line (Figure 1a). Since applying more than 14 µM of cisplatin to the normal cell is highly toxic, in this study, a combination of chemotherapy and gene therapy strategies was applied to assess the possibility of eradicating the cisplatin-resistant A549 cell line, using ferroptosis or apoptosis induction.
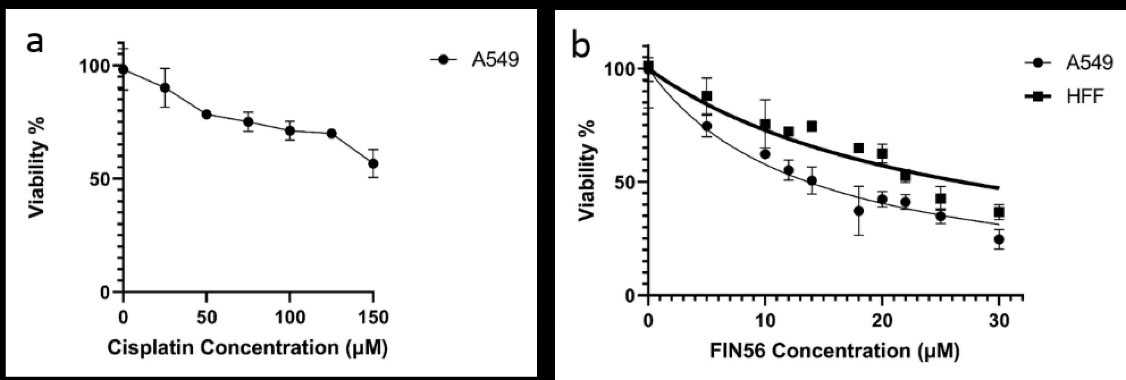
Figure 1.
Cisplatin-resistant A549 cells’ sensitivity to cisplatin (a) and FIN56 (b) were evaluated by MTT assay. Cancerous A549 has higher sensitivity to FIN56 than normal HFF cells.
.
Cisplatin-resistant A549 cells’ sensitivity to cisplatin (a) and FIN56 (b) were evaluated by MTT assay. Cancerous A549 has higher sensitivity to FIN56 than normal HFF cells.
Ferroptosis is a ROS-dependent and caspase-independent cell death pathway, which is naturally applied by cytotoxic killer cells to eradicate tumor cells.73,74 In ferroptosis, usually cellular lipid peroxidation levels are augmented due to the accumulation of ROS molecules. The excessive cellular ROS production in ferroptosis is related to the malfunction of mitochondrial membrane potential, cellular thiol-dependent antioxidant system malfunction, and cellular antioxidant regulatory pathways such as mevalonate and Nrf2 pathways.75,76
Notably, ROS plays diverse roles in cancer cell’s fate. Based on the cancer cells’ distinct metabolism and tumor hypoxia, the cancer cells have higher ROS concentrations than normal cells. The increased ROS level can induce DNA mutagenesis and help the heterogenicity of the tumors. Moreover, ROS molecules’ continuous exposure can persuade cell proliferation by activating oncogenic proteins, including growth factor receptors, VEGF, Ras, MAPK, and PI3K/AKT. Furthermore, ROS molecules elevate the activity of Nrf2, FOXO, and HIF1α antioxidant transcription factors and subsequent antioxidant enzymes in cancer cells. This procedure led to a higher level of the cellular antioxidant system such as Heme oxygenase, glutathione peroxidase (GPX), superoxide dismutase, catalase, and glutathione (GSH) in cancer cells.77
However, as most chemotherapeutic drugs are ROS generators, these compounds’ administration can alter the cancer cells’ redox homeostasis. The elevated ROS concentrations suppress regular cellular enzymatic activity and cell connectivity, cause DNA damage, and cease cell cycle progression. ROS, while at higher concentrations, impairing the cells’ physiological function, results in a distinct type of regulated cell death such as ferroptosis.78
Therefore, ROS-dependent cell death strategies such as ferroptosis have been used in cancer therapy, especially for eradicating the resistant forms of cancers.77,79
FIN56 and GPX4 siRNA induce ferroptosis in A549 cisplatin-resistant cells
In this study, two ferroptosis inducers, FIN56 small molecule and, GPX4-siRNA were harnessed in the battle against cisplatin-resistant A549 cells. We used the MTT assay to determine the cytotoxicity of FIN56 in both cancerous and normal cell lines. As shown in (Figure 1b), both cell lines’ viability was reduced after 48 hours incubation with FIN56 in a dose-dependent manner. However, the IC50 value of FIN56 for A549 and HFF was calculated as 12.71 and 24.97 μM, respectively. Therefore, these results indicate that resistant A549 cells are more sensitive to FIN56 than the HFF normal cells. Additionally, as 5µM of the FIN56 is sufficient for inducing the ferroptosis, and the normal cells have an upper than 80% viability (Figure 1b), this concentration opted for the rest of this study.
Along with the chemical induction of ferroptosis, GPX4 siRNA gene silencing was also performed. To determine the efficacy of gene silencing, we performed the western blot analysis after 48 hours of siRNA incubation. We observed that GPX4 protein’s quantity decreased by 35%, calculated using ImageJ software, compared to their control groups, as shown in (Figure 2).
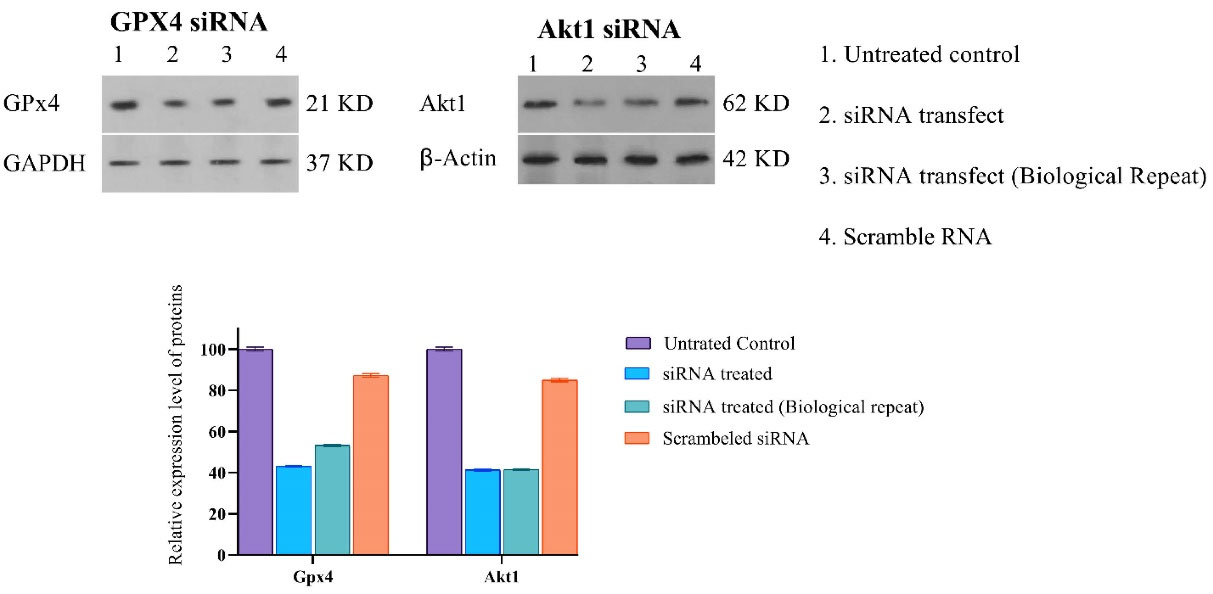
Figure 2.
Effects of Akt1 and GPX4 siRNA on the expression of GPX4 and AKT1 in A549 cells. siRNAs were added to the A549 cell culture for 48 h, and cells were harvested to be processed for Western blot analysis. (A) Protein bands were shown as above, Lane 1: control; lanes 2&3: 1ng Akt1 siRNA; lane 4: scramble RNA. (B) Viability of A549 cell line after treatment by siRNAs for 48h
.
Effects of Akt1 and GPX4 siRNA on the expression of GPX4 and AKT1 in A549 cells. siRNAs were added to the A549 cell culture for 48 h, and cells were harvested to be processed for Western blot analysis. (A) Protein bands were shown as above, Lane 1: control; lanes 2&3: 1ng Akt1 siRNA; lane 4: scramble RNA. (B) Viability of A549 cell line after treatment by siRNAs for 48h
The cellular accumulation of ROS molecules is considered hallmarks of ferroptosis induction. The emission of fluorescent dyes DCFDA and BODIPY-C11 was assessed by flow-cytometry to confirm the ferroptosis induction in FIN56 and GPX4 siRNA-treated A549 cells. As shown in (Figure 3), the total and lipid ROS amount were shifted to higher signals in FIN56 (5μM), and GPX4 siRNA (100nM) treated cells (48 hours) compared to their control groups, which indicates successful ferroptosis induction in both treatments.
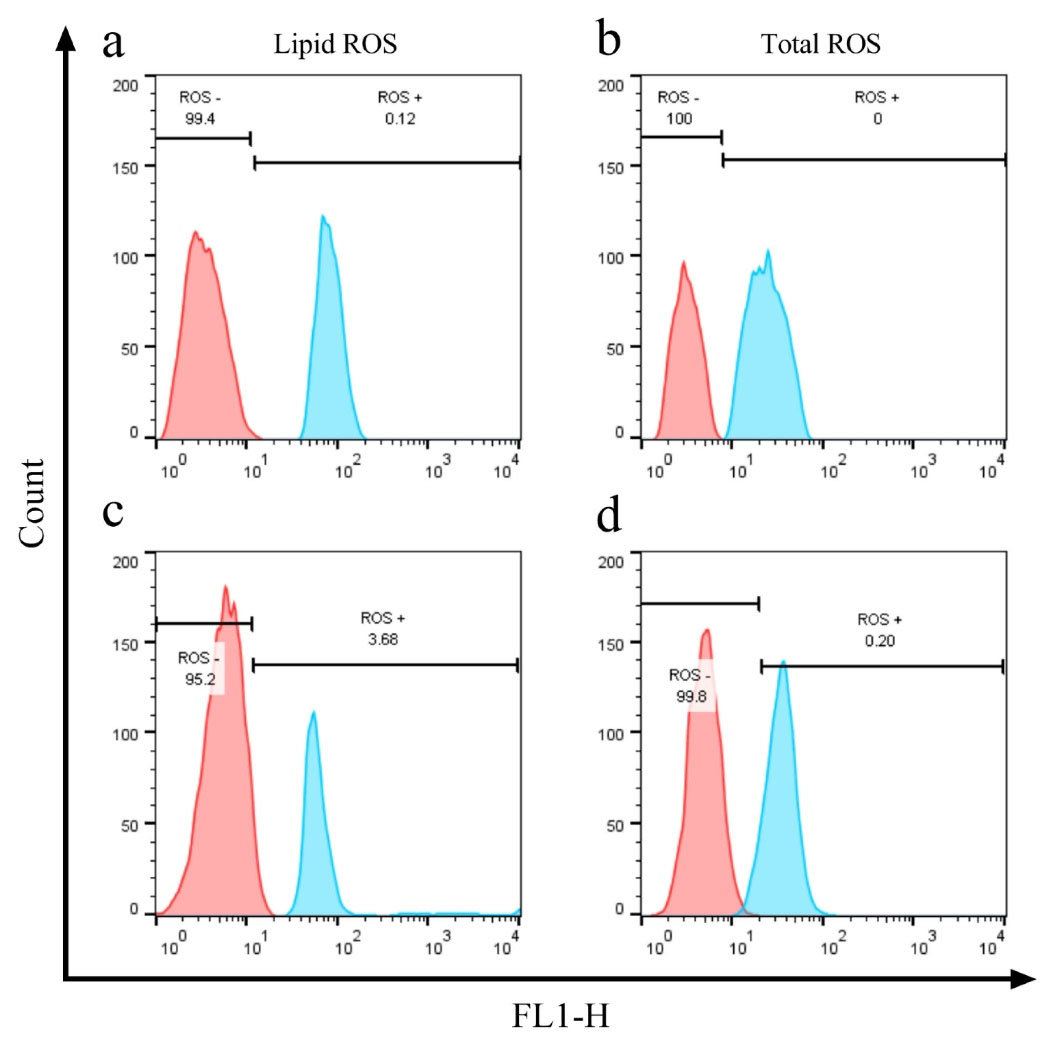
Figure 3.
Effect of FIN 56 (Panels a and b) and GPX4 siRNA (Panels c and d) on the cellular lipid and total ROS levels. Cells were treated with 5μM FIN56 and 1 ng siRNA for 48 h. DCFDA and BODIPY-C11, respectively, measured total ROS and lipid ROS levels
.
Effect of FIN 56 (Panels a and b) and GPX4 siRNA (Panels c and d) on the cellular lipid and total ROS levels. Cells were treated with 5μM FIN56 and 1 ng siRNA for 48 h. DCFDA and BODIPY-C11, respectively, measured total ROS and lipid ROS levels
Akt1 siRNA induce ferroptosis in A549 cisplatin-resistant cells
There are numerous reports on the role of the Akt and its cooperator proteins such as PI3K, mTOR, NF-κB, c-Met, c-Myc, and ERK1/2 in cisplatin resistance induction in the A549 cell line.80-83 These signaling pathways alter apoptotic (Bax, Bad, Bim) and anti-apoptotic (Bcl2, Bcl-xl) gene expression levels, inhibiting apoptosis induction in these resistance cells. Therefore, Akt-related pathways have been the center of attention in several chemoresistances re-sensitization studies.84-88
Based on previous in vitro reports, administration of vinorelbine, sunitinib, BAICALEIN, or genistein alleviates the cytotoxicity of cisplatin in the resistance of the A549 cell line through inhibition of the Akt pathway and other cisplatin resistance-related mechanisms such as drug metabolism, efflux, and DNA repair machinery.52,89-91
Correspondingly, gene therapy strategies also have been applied to overcome chemoresistance in cisplatin-based therapies. Replacement gene therapy of tumor suppressor genes such as PTEN,92 IL-2493,94 re-sensitize the cisplatin-resistant A549 cells via downregulation of the PI3K/AKT/hTERT pathway. Similarly, knocking down the overexpressed aldehyde dehydrogenase 1A1,95 Tripartite motif-containing 59 oncogene protein,96 and MDR197 restore the cisplatin toxicity in the resistance A549 cell line in an Akt dependent manner.
Similarly, in this study, silencing the Akt1 gene expression using the 100 nM, specific siRNA was opted to eradicate the cisplatin-resistant A549 cells. The western blot analysis after 48 hours of siRNA incubation indicates that total amounts of Akt1 protein were successfully downregulated distinctively to 41% (Figure 2).
Correspondingly, the flow-cytometry technique’s shift of Annexin V/PI stained cells revealed that Akt1 siRNA increased the percentage of late apoptotic cells from 0.75% to 48.1% and early apoptotic cells from 1.48% to 30.5% meaningfully (Figure 4). Therefore, the Akt1 siRNA effectively induces apoptosis in the A549 resistant cells.
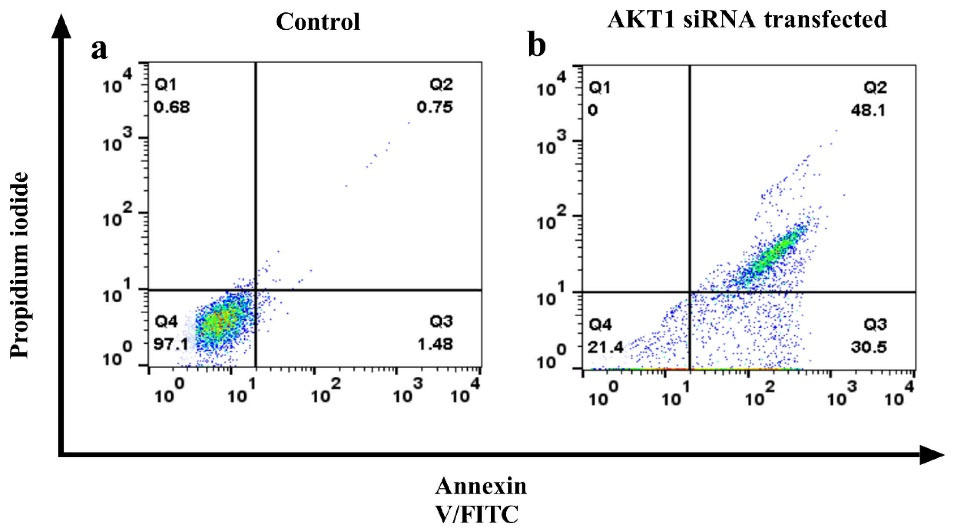
Figure 4.
Flow-cytometry analysis of treated Akt1 siRNA and the control group. The panels are shown in the following order: (a) control group; (b) Akt1 siRNA treated group. Dots with Annexin V−/PI + (Q1), Annexin V + /PI + (Q2), Annexin V + /PI− (Q3), and Annexin V−/PI− (Q4) and features represent necrotically, late apoptotic, early apoptotic, and viable intact cells, respectively
.
Flow-cytometry analysis of treated Akt1 siRNA and the control group. The panels are shown in the following order: (a) control group; (b) Akt1 siRNA treated group. Dots with Annexin V−/PI + (Q1), Annexin V + /PI + (Q2), Annexin V + /PI− (Q3), and Annexin V−/PI− (Q4) and features represent necrotically, late apoptotic, early apoptotic, and viable intact cells, respectively
Antioxidant related genes were down-regulated after FIN56 and siRNA treatments
Based on the previous knowledge about ferroptosis and its inducers, the ROS accumulation in the cells is negatively correlated with the level and activity of the antioxidant gene regulator (NRF2 and AhR), antioxidant enzymes (GPX4), and antioxidant molecules (CoQ10, glutathione).73,98,99 Mainly, ferroptosis inducers disturb the function of this antioxidant redox balance regulator system.100 However, these antioxidant systems’ elevated levels and activity can induce resistance against ferroptosis in a cell-type-specific manner.101,102
Correspondingly, this study evaluated the expression variation of anti-ferroptosis and anti-apoptosis-related genes, mainly GPX4, CoQ10, Nerf2, and Akt1 genes, using a real-time PCR technique. The graphical representation of different treatments’ gene expression ratios presents in (Figure 5). Considering the ferroptosis induction, after the treatment of FIN56, the gene expression ratio of GPX4, CoQ10, and Nerf2 was calculated as 0.0216, 0.00059, and 0.05 compared to the non-treated control group. While following the transfection of GPX4 siRNA, the gene expression ratio of the mentioned genes was 0.02, 0.039, and 0.69, respectively. The results reveal that following either GPX4-siRNA or FIN56 treatments, the expression level of anti-ferroptosis Nrf2, GPX4, and CoQ10 genes was significantly declined (P < 0.001) compared to the correspondence untreated control group. Likewise, for apoptosis induction, the Akt1 gene expression ratio in Akt1 siRNA-treated A549 cells was 0.0138, indicating effective inhibition of the Akt1 expression.
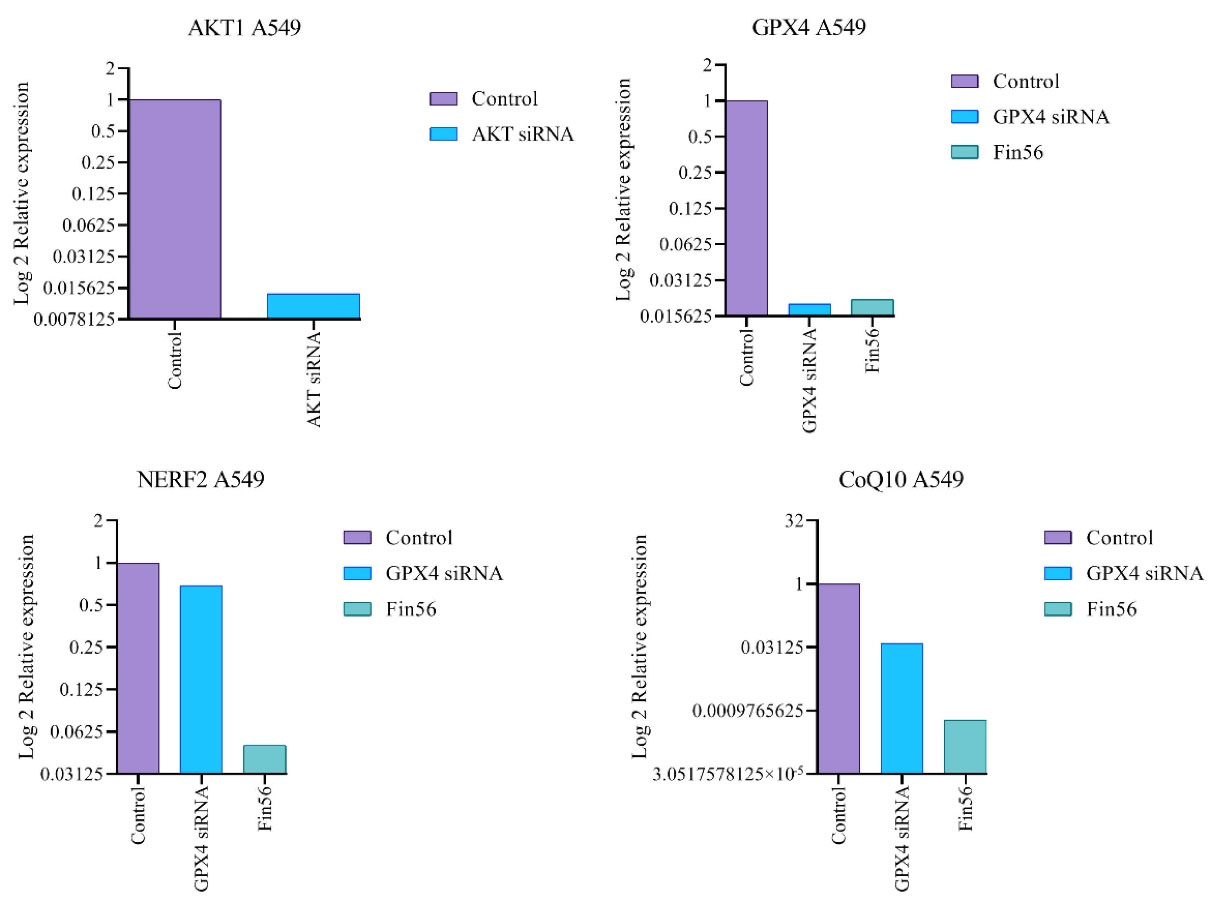
Figure 5.
Akt1, GPX4, CoQ10, and, Nerf2 gene expression ratios were obtained by real-time PCR with treatments (Akt1 and GPX4 siRNAs separately and FIN56) in resistant A549 cell lines. Target genes were normalized to GAPDH as a control gene
.
Akt1, GPX4, CoQ10, and, Nerf2 gene expression ratios were obtained by real-time PCR with treatments (Akt1 and GPX4 siRNAs separately and FIN56) in resistant A549 cell lines. Target genes were normalized to GAPDH as a control gene
FIN56, along with GPX4 and Akt1 siRNAs, destroy A549 cisplatin-resistant cells
The results of this indicated cell viability alterations in combination therapy of “Cisplatin combined by ferroptosis or apoptosis inducers” in A549 resistant cells by Trypan blue exclusion dye assay, using the Flow cytometry technique. As shown in (Figure 6), the highest percentage of dead cells was observed after treatment with “Cisplatin + 5μM FIN56” at 92%. Meanwhile, administration of either “Cisplatin + 100 ng Gpx4 siRNA” or “Cisplatin + 100 ng Akt1 siRNA” results in 85% dead cells compared to the control group.
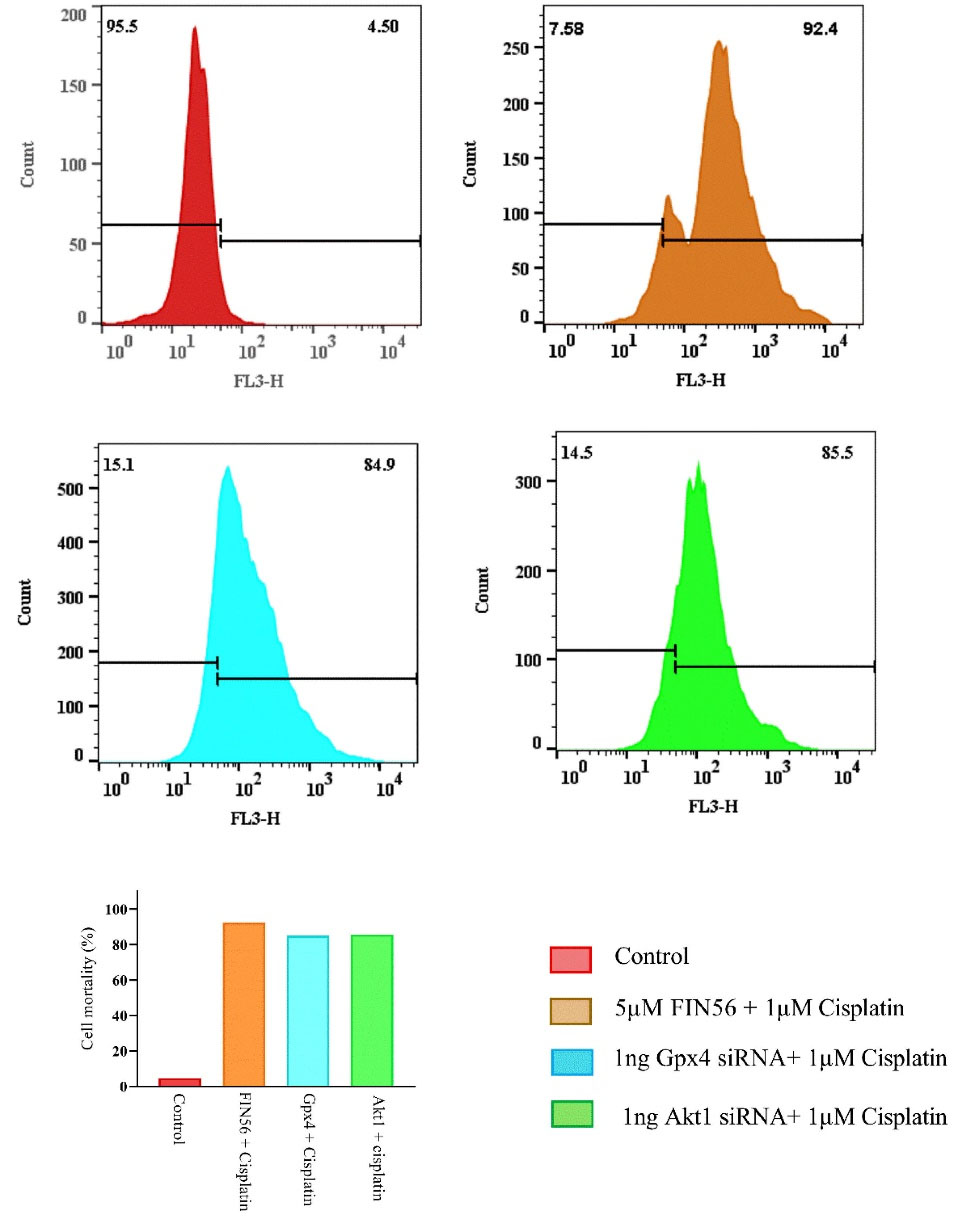
Figure 6.
Histogram analysis of the trypan blue exclusion assay by flow-cytometry to evaluate cells’ mortality after treating by 5μM FIN56 + Cisplatin, 1 ng Akt1 siRNA + Cisplatin, 1 ng Gpx4 siRNA + cisplatin compared with the control group
.
Histogram analysis of the trypan blue exclusion assay by flow-cytometry to evaluate cells’ mortality after treating by 5μM FIN56 + Cisplatin, 1 ng Akt1 siRNA + Cisplatin, 1 ng Gpx4 siRNA + cisplatin compared with the control group
Similarly, several reports from Roh and colleagues highlight the ferroptosis cell death strategy’s impact on eradicating the cisplatin-resistant cancer models.16,103 They showed that Erastin and Sulfasalazine could be used to overcome cisplatin resistance through ferroptosis induction. However, another ferroptosis inducer, like RSL3 activity against the same cisplatin-resistant cell line, depends on the NRF2 pathway activity.15 Similarly, RNA sequencing data of the Erastin sensitive and resistant cell lines reveals that the transcriptional activity of NRF2 and AhR is one of the most critical factors in the ferroptosis-related resistance phenotype. Principally, it was shown that A549 cells, as an epithelial lung cancer model, have NRF2 and AhR mediated resistance to Erastin. However, A549 transdifferentiated mesenchymal lung cancer cells, which have chemoresistance to some therapeutics, are sensitive to erastin.104
This difference in the A549 ferroptosis sensitivity may also be related to the mechanism of ferroptosis inducers. Erastin and Sulfasalazine are Cystine-Glutamate-antiporter inhibitors that lead to cellular glutathione depletion. However, RSL3 is a GPX4 antagonist, and FIN56, like siRNA, depletes cellular GPX4 protein, leading to lipid ROS accumulation.21
Previous studies show that directly targeting the Akt using siRNA can induce apoptosis in resistance cells.105,106 Similarly, in this study, based on the Trypan blue exclusion assay and Annexin-PI staining flow cytometry results, Akt1 downregulation using the siRNA induced about 80% programed apoptosis death. This outcome might be related to the down-regulation of the Akt1-dependent several anti-apoptotic proteins such as the Bad and Bcl families.107
Conclusion
In conclusion, chemotherapy resistance NSCLC cells could be eradicated either by reinforcing the apoptosis by targeting the Akt1 as a critical cellular survival regulator; or by disrupting the cellular ROS homeostasis using ferroptosis inducers. Therefore, Akt1 or GPX4 siRNA combined with drug administration could be considered a promising strategy in NSCLC therapy.
Acknowledgments
This project is part of a Ph.D. thesis (grant No. 59305) funded by Drug Applied Research Center, Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran.
Author Contributions
Conceptualization: Ahmad Yari Khosroushahi.
Data curation: Morteza Golbashirzadeh.
Formal Analysis: Morteza Golbashirzadeh.
Funding acquisition: Hamid Reza Heidari.
Investigation: Morteza Golbashirzadeh.
Methodology: Ahmad Yari Khosroushahi,, Mehdi Talebi.
Project administration: Hamid Reza Heidari.
Resources: Ahmad Yari Khosroushahi.
Supervision: Ahmad Yari Khosroushahi.
Validation: Hamid Reza Heidari.
Visualization: Mehdi Talebi.
Writing – original draft: Morteza Golbashirzadeh.
Writing – review & editing: Hamid Reza Heidari.
Ethical Issues
Not applicable.
Conflict of Interest
The authors declare no conflict of interest.
References
- Portal D, Hofstetter L, Eshed I, Dan-Lantsman C, Sella T, Urban D. L3 skeletal muscle index (L3SMI) is a surrogate marker of sarcopenia and frailty in non-small cell lung cancer patients. Cancer Manag Res 2019; 11:2579-88. doi: 10.2147/cmar.s195869 [Crossref] [ Google Scholar]
- Planchard D, Popat S, Kerr K, Novello S, Smit EF, Faivre-Finn C. Correction to: “Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up”. Ann Oncol 2019; 30(5):863-70. doi: 10.1093/annonc/mdy474 [Crossref] [ Google Scholar]
- Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med 2005; 353(2):123-32. doi: 10.1056/NEJMoa050753 [Crossref] [ Google Scholar]
- Cai X, Hu B, Liu S, Liu M, Huang Y, Lei P, et al. Close homologue of L1 sensitizes lung cancer cells to cisplatin and paclitaxel via inhibition Akt pathway. bioRxiv [Preprint]. August 28, 2019. Available from: https://www.biorxiv.org/content/10.1101/747238v1.
- Park KS, Kim HK, Lee JH, Choi YB, Park SY, Yang SH. Transglutaminase 2 as a cisplatin resistance marker in non-small cell lung cancer. J Cancer Res Clin Oncol 2010; 136(4):493-502. doi: 10.1007/s00432-009-0681-6 [Crossref] [ Google Scholar]
- Nascimento AV, Singh A, Bousbaa H, Ferreira D, Sarmento B, Amiji MM. Overcoming cisplatin resistance in non-small cell lung cancer with Mad2 silencing siRNA delivered systemically using EGFR-targeted chitosan nanoparticles. Acta Biomater 2017; 47:71-80. doi: 10.1016/j.actbio.2016.09.045 [Crossref] [ Google Scholar]
- Jiang GB, Fang HY, Tao DY, Chen XP, Cao FL. COX-2 potentiates cisplatin resistance of non-small cell lung cancer cells by promoting EMT in an AKT signaling pathway-dependent manner. Eur Rev Med Pharmacol Sci 2019; 23(9):3838-46. doi: 10.26355/eurrev_201905_17811 [Crossref] [ Google Scholar]
- Teng X, Fan XF, Li Q, Liu S, Wu DY, Wang SY. XPC inhibition rescues cisplatin resistance via the Akt/mTOR signaling pathway in A549/DDP lung adenocarcinoma cells. Oncol Rep 2019; 41(3):1875-82. doi: 10.3892/or.2019.6959 [Crossref] [ Google Scholar]
- Cruz-Bermúdez A, Laza-Briviesca R, Vicente-Blanco RJ, García-Grande A, Coronado MJ, Laine-Menéndez S. Cisplatin resistance involves a metabolic reprogramming through ROS and PGC-1α in NSCLC which can be overcome by OXPHOS inhibition. Free Radic Biol Med 2019; 135:167-81. doi: 10.1016/j.freeradbiomed.2019.03.009 [Crossref] [ Google Scholar]
- Galluzzi L, Vitale I, Michels J, et al. Systems biology of cisplatin resistance: past, present and future. Cell Death Dis 2014;5(5):e1257. Published 2014 May 29. 10.1038/cddis.2013.428.
- Peng G, Tang Z, Xiang Y, Chen W. Glutathione peroxidase 4 maintains a stemness phenotype, oxidative homeostasis and regulates biological processes in Panc-1 cancer stem-like cells. Oncol Rep 2019; 41(2):1264-74. doi: 10.3892/or.2018.6905 [Crossref] [ Google Scholar]
- Sun DM, Tang BF, Li ZX, Guo HB, Cheng JL, Song PP. MiR-29c reduces the cisplatin resistance of non-small cell lung cancer cells by negatively regulating the PI3K/Akt pathway. Sci Rep 2018; 8(1):8007. doi: 10.1038/s41598-018-26381-w [Crossref] [ Google Scholar]
- Wang W, Zhang Y, Chen R, Tian Z, Zhai Y, Janz S. Chromosomal instability and acquired drug resistance in multiple myeloma. Oncotarget 2017; 8(44):78234-44. doi: 10.18632/oncotarget.20829 [Crossref] [ Google Scholar]
- Yang X, Zhang Q, Yang X, Zhao M, Yang T, Yao A. PACT cessation overcomes ovarian cancer cell chemoresistance to cisplatin by enhancing p53-mediated apoptotic pathway. BiochemBiophys Res Commun 2019; 511(4):719-24. doi: 10.1016/j.bbrc.2019.02.089 [Crossref] [ Google Scholar]
- Shin D, Kim EH, Lee J, Roh JL. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic Biol Med 2018; 129:454-62. doi: 10.1016/j.freeradbiomed.2018.10.426 [Crossref] [ Google Scholar]
- Roh JL, Kim EH, Jang HJ, Park JY, Shin D. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett 2016; 381(1):96-103. doi: 10.1016/j.canlet.2016.07.035 [Crossref] [ Google Scholar]
- Sajadimajd S, Khazaei M. Oxidative stress and cancer: the role of Nrf2. Curr Cancer Drug Targets 2018; 18(6):538-57. doi: 10.2174/1568009617666171002144228 [Crossref] [ Google Scholar]
- Sadhukhan P, Saha S, Sil P. Targeting oxidative stress: a novel approach in mitigating cancer. Biochem Anal Biochem 2015; 4(4):236. doi: 10.4172/2161-1009.1000236 [Crossref] [ Google Scholar]
- Feng M, Zhong LX, Zhan ZY, Huang ZH, Xiong JP. Enhanced antitumor efficacy of resveratrol-loaded nanocapsules in colon cancer cells: physicochemical and biological characterization. Eur Rev Med Pharmacol Sci 2017; 21(2):375-82. [ Google Scholar]
- Bai Y, Meng L, Han L, Jia Y, Zhao Y, Gao H. Lipid storage and lipophagy regulates ferroptosis. BiochemBiophys Res Commun 2019; 508(4):997-1003. doi: 10.1016/j.bbrc.2018.12.039 [Crossref] [ Google Scholar]
- Feng H, Stockwell BR. Unsolved mysteries: How does lipid peroxidation cause ferroptosis?. PLoS Biol 2018; 16(5):e2006203. doi: 10.1371/journal.pbio.2006203 [Crossref] [ Google Scholar]
- Dixon SJ. Ferroptosis: bug or feature?. Immunol Rev 2017; 277(1):150-7. doi: 10.1111/imr.12533 [Crossref] [ Google Scholar]
- Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X. Ferroptosis: process and function. Cell Death Differ 2016; 23(3):369-79. doi: 10.1038/cdd.2015.158 [Crossref] [ Google Scholar]
- Cui Q, Wang JQ, Assaraf YG, Ren L, Gupta P, Wei L. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist Updat 2018; 41:1-25. doi: 10.1016/j.drup.2018.11.001 [Crossref] [ Google Scholar]
- Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med 2019; 133:144-52. doi: 10.1016/j.freeradbiomed.2018.09.014 [Crossref] [ Google Scholar]
- Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK. CD8 + T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019; 569(7755):270-4. doi: 10.1038/s41586-019-1170-y [Crossref] [ Google Scholar]
- Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N. Ferroptosis: past, present and future. Cell Death Dis 2020; 11(2):88. doi: 10.1038/s41419-020-2298-2 [Crossref] [ Google Scholar]
- Lei P, Bai T, Sun Y. Mechanisms of ferroptosis and relations with regulated cell death: a review. Front Physiol 2019; 10:139. doi: 10.3389/fphys.2019.00139 [Crossref] [ Google Scholar]
- Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 2014; 16(12):1180-91. doi: 10.1038/ncb3064 [Crossref] [ Google Scholar]
- Park TJ, Park JH, Lee GS, Lee JY, Shin JH, Kim MW. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis 2019; 10(11):835. doi: 10.1038/s41419-019-2061-8 [Crossref] [ Google Scholar]
- Zhang Z, Wu Y, Yuan S, Zhang P, Zhang J, Li H. Glutathione peroxidase 4 participates in secondary brain injury through mediating ferroptosis in a rat model of intracerebral hemorrhage. Brain Res 2018; 1701:112-25. doi: 10.1016/j.brainres.2018.09.012 [Crossref] [ Google Scholar]
- Su Y, Zhao B, Zhou L, Zhang Z, Shen Y, Lv H. Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer Lett 2020; 483:127-36. doi: 10.1016/j.canlet.2020.02.015 [Crossref] [ Google Scholar]
- Ye Z, Liu W, Zhuo Q, Hu Q, Liu M, Sun Q. Ferroptosis: final destination for cancer?. Cell Prolif 2020; 53(3):e12761. doi: 10.1111/cpr.12761 [Crossref] [ Google Scholar]
- Sugiyama A, Ohta T, Obata M, Takahashi K, Seino M, Nagase S. xCT inhibitor sulfasalazine depletes paclitaxel-resistant tumor cells through ferroptosis in uterine serous carcinoma. Oncol Lett 2020; 20(3):2689-700. doi: 10.3892/ol.2020.11813 [Crossref] [ Google Scholar]
- Steckiewicz KP, Barcinska E, Malankowska A, Zauszkiewicz-Pawlak A, Nowaczyk G, Zaleska-Medynska A. Impact of gold nanoparticles shape on their cytotoxicity against human osteoblast and osteosarcoma in in vitro model. Evaluation of the safety of use and anti-cancer potential. J Mater Sci Mater Med 2019; 30(2):22. doi: 10.1007/s10856-019-6221-2 [Crossref] [ Google Scholar]
- Vakili Saatloo M, Aghbali A, Koohsoltani M, Yari Khosroushahi A. Akt1 and Jak1 siRNA based silencing effects on the proliferation and apoptosis in head and neck squamous cell carcinoma. Gene 2019; 714:143997. doi: 10.1016/j.gene.2019.143997 [Crossref] [ Google Scholar]
- Basaki Y, Hosoi F, Oda Y, Fotovati A, Maruyama Y, Oie S. Akt-dependent nuclear localization of Y-box-binding protein 1 in acquisition of malignant characteristics by human ovarian cancer cells. Oncogene 2007; 26(19):2736-46. doi: 10.1038/sj.onc.1210084 [Crossref] [ Google Scholar]
- Vandermoere F, El Yazidi-Belkoura I, Slomianny C, Demont Y, Bidaux G, Adriaenssens E. The valosin-containing protein (VCP) is a target of Akt signaling required for cell survival. J Biol Chem 2006; 281(20):14307-13. doi: 10.1074/jbc.M510003200 [Crossref] [ Google Scholar]
- Hsieh AC, Bo R, Manola J, Vazquez F, Bare O, Khvorova A. A library of siRNA duplexes targeting the phosphoinositide 3-kinase pathway: determinants of gene silencing for use in cell-based screens. Nucleic Acids Res 2004; 32(3):893-901. doi: 10.1093/nar/gkh238 [Crossref] [ Google Scholar]
- Pan X, Lin Z, Jiang D, Yu Y, Yang D, Zhou H. Erastin decreases radioresistance of NSCLC cells partially by inducing GPX4-mediated ferroptosis. Oncol Lett 2019; 17(3):3001-8. doi: 10.3892/ol.2019.9888 [Crossref] [ Google Scholar]
- NaveenKumar SK, Hemshekhar M, Kemparaju K, Girish KS. Hemin-induced platelet activation and ferroptosis is mediated through ROS-driven proteasomal activity and inflammasome activation: protection by melatonin. BiochimBiophys Acta Mol Basis Dis 2019; 1865(9):2303-16. doi: 10.1016/j.bbadis.2019.05.009 [Crossref] [ Google Scholar]
- Wang C, Cui C. Inhibition of lung cancer proliferation by wogonin is associated with activation of apoptosis and generation of reactive oxygen species. Balkan Med J 2019; 37(1):29-33. doi: 10.4274/balkanmedj.galenos.2019.2019.7.75 [Crossref] [ Google Scholar]
- Sun X, Niu X, Chen R, He W, Chen D, Kang R. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology 2016; 64(2):488-500. doi: 10.1002/hep.28574 [Crossref] [ Google Scholar]
- Sangeeta K, Yenugu S. siRNA-mediated knockdown of sperm-associated antigen 11a (Spag11a) mRNA in epididymal primary epithelial cells affects proliferation: a transcriptome analyses. Cell Tissue Res 2020; 379(3):601-12. doi: 10.1007/s00441-019-03107-6 [Crossref] [ Google Scholar]
- Liang Y, Zhang H, Song X, Yang Q. Metastatic heterogeneity of breast cancer: molecular mechanism and potential therapeutic targets. Semin Cancer Biol 2020; 60:14-27. doi: 10.1016/j.semcancer.2019.08.012 [Crossref] [ Google Scholar]
- Wu Z, Fournel L, Stadler N, Liu J, Boullier A, Hoyeau N. Modulation of lung cancer cell plasticity and heterogeneity with the restoration of cisplatin sensitivity by neurotensin antibody. Cancer Lett 2019; 444:147-61. doi: 10.1016/j.canlet.2018.12.007 [Crossref] [ Google Scholar]
- Sun L, Li YY, Ma JT, Zhang SL, Huang LT, Han CB. The influence of tumor heterogeneity on sensitivity of EGFR-mutant lung adenocarcinoma cells to EGFR-TKIs. Transl Cancer Res 2019; 8(5):1834-44. doi: 10.21037/tcr.2019.09.01 [Crossref] [ Google Scholar]
- Lim ZF, Ma PC. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J Hematol Oncol 2019; 12(1):134. doi: 10.1186/s13045-019-0818-2 [Crossref] [ Google Scholar]
- Shue YT, Lim JS, Sage J. Tumor heterogeneity in small cell lung cancer defined and investigated in pre-clinical mouse models. Transl Lung Cancer Res 2018; 7(1):21-31. doi: 10.21037/tlcr.2018.01.15 [Crossref] [ Google Scholar]
- Niehr F, Eder T, Pilz T, Konschak R, Treue D, Klauschen F. Multilayered omics-based analysis of a head and neck cancer model of cisplatin resistance reveals intratumoral heterogeneity and treatment-induced clonal selection. Clin Cancer Res 2018; 24(1):158-68. doi: 10.1158/1078-0432.ccr-17-2410 [Crossref] [ Google Scholar]
- Wang L, Zhu B, Zhang M, Wang X. Roles of immune microenvironment heterogeneity in therapy-associated biomarkers in lung cancer. Semin Cell Dev Biol 2017; 64:90-7. doi: 10.1016/j.semcdb.2016.09.008 [Crossref] [ Google Scholar]
- Liu D, Yan L, Wang L, Tai W, Wang W, Yang C. Genistein enhances the effect of cisplatin on the inhibition of non-small cell lung cancer A549 cell growth in vitro and in vivo. Oncol Lett 2014; 8(6):2806-10. doi: 10.3892/ol.2014.2597 [Crossref] [ Google Scholar]
- Wang Y, Liu L, Chen Z. Transcriptome profiling of cervical cancer cells acquired resistance to cisplatin by deep sequencing. Artif Cells NanomedBiotechnol 2019; 47(1):2820-9. doi: 10.1080/21691401.2019.1637882 [Crossref] [ Google Scholar]
- Zhou Y, Wang SY, Yang XK, Wang HM. [Relationship between the Akt-regulated direct p53 mitochondrial translocation and the resistance to cisplatin of ovarian cancer cells]. Zhonghua Zhong Liu Za Zhi 2011; 33(2):97-100. doi: 10.3760/cma.j.issn.0253-3766.2011.02.004 [Crossref] [ Google Scholar]
- Suzuki E, Daniels TR, Helguera G, Penichet ML, Umezawa K, Bonavida B. Inhibition of NF-kappaB and Akt pathways by an antibody-avidin fusion protein sensitizes malignant B-cells to cisplatin-induced apoptosis. Int J Oncol 2010; 36(5):1299-307. doi: 10.3892/ijo_00000615 [Crossref] [ Google Scholar]
- Shimamura T, Li D, Ji H, Haringsma HJ, Liniker E, Borgman CL. Hsp90 inhibition suppresses mutant EGFR-T790M signaling and overcomes kinase inhibitor resistance. Cancer Res 2008; 68(14):5827-38. doi: 10.1158/0008-5472.can-07-5428 [Crossref] [ Google Scholar]
- Giopanou I, Pintzas A. RAS and BRAF in the foreground for non-small cell lung cancer and colorectal cancer: similarities and main differences for prognosis and therapies. Crit Rev Oncol Hematol 2020; 146:102859. doi: 10.1016/j.critrevonc.2019.102859 [Crossref] [ Google Scholar]
- Lin JJ, Riely GJ, Shaw AT. Targeting ALK: precision medicine takes on drug resistance. Cancer Discov 2017; 7(2):137-55. doi: 10.1158/2159-8290.cd-16-1123 [Crossref] [ Google Scholar]
- Moro M, Caiola E, Ganzinelli M, Zulato E, Rulli E, Marabese M. Metformin enhances cisplatin-induced apoptosis and prevents resistance to cisplatin in co-mutated KRAS/LKB1 NSCLC. J Thorac Oncol 2018; 13(11):1692-704. doi: 10.1016/j.jtho.2018.07.102 [Crossref] [ Google Scholar]
- Peng DH, Rodriguez BL, Diao L, Chen L, Wang J, Byers LA. Collagen promotes anti-PD-1/PD-L1 resistance in cancer through LAIR1-dependent CD8 + T cell exhaustion. Nat Commun 2020; 11(1):4520. doi: 10.1038/s41467-020-18298-8 [Crossref] [ Google Scholar]
- Meng F, Wang F, Wang L, Wong SC, Cho WC, Chan LW. MiR-30a-5p overexpression may overcome EGFR-inhibitor resistance through regulating PI3K/AKT signaling pathway in non-small cell lung cancer cell lines. Front Genet 2016; 7:197. doi: 10.3389/fgene.2016.00197 [Crossref] [ Google Scholar]
- Sun CY, Zhu Y, Li XF, Wang XQ, Tang LP, Su ZQ. Scutellarin increases cisplatin-induced apoptosis and autophagy to overcome cisplatin resistance in non-small cell lung cancer via ERK/p53 and c-met/AKT signaling pathways. Front Pharmacol 2018; 9:92. doi: 10.3389/fphar.2018.00092 [Crossref] [ Google Scholar]
- Fujimoto Y, Morita TY, Ohashi A, Haeno H, Hakozaki Y, Fujii M. Combination treatment with a PI3K/Akt/mTOR pathway inhibitor overcomes resistance to anti-HER2 therapy in PIK3CA-mutant HER2-positive breast cancer cells. Sci Rep 2020; 10(1):21762. doi: 10.1038/s41598-020-78646-y [Crossref] [ Google Scholar]
- Daly C, Castanaro C, Zhang W, Zhang Q, Wei Y, Ni M. FGFR3-TACC3 fusion proteins act as naturally occurring drivers of tumor resistance by functionally substituting for EGFR/ERK signaling. Oncogene 2017; 36(4):471-81. doi: 10.1038/onc.2016.216 [Crossref] [ Google Scholar]
- Li Y, Jia L, Liu C, Gong Y, Ren D, Wang N. Axl as a downstream effector of TGF-β1 via PI3K/Akt-PAK1 signaling pathway promotes tumor invasion and chemoresistance in breast carcinoma. Tumour Biol 2015; 36(2):1115-27. doi: 10.1007/s13277-014-2677-3 [Crossref] [ Google Scholar]
- Jiang W, Xia J, Xie S, Zou R, Pan S, Wang ZW. Long non-coding RNAs as a determinant of cancer drug resistance: towards the overcoming of chemoresistance via modulation of lncRNAs. Drug Resist Updat 2020; 50:100683. doi: 10.1016/j.drup.2020.100683 [Crossref] [ Google Scholar]
- Yousefi H, Maheronnaghsh M, Molaei F, Mashouri L, Aref AR, Momeny M. Long noncoding RNAs and exosomal lncRNAs: classification, and mechanisms in breast cancer metastasis and drug resistance. Oncogene 2020; 39(5):953-74. doi: 10.1038/s41388-019-1040-y [Crossref] [ Google Scholar]
- Bahannan AH, Johnson T, Myles E. Comparison of violacein, mangosteen and sidr extracts as agents for inhibiting breast and lung cancer cell growth. FASEB J 2017; 31(Suppl 1):lb168. doi: 10.1096/fasebj.31.1_supplement.lb168 [Crossref] [ Google Scholar]
- Yang CP, Verdier-Pinard P, Wang F, Lippaine-Horvath E, He L, Li D. A highly epothilone B-resistant A549 cell line with mutations in tubulin that confer drug dependence. Mol Cancer Ther 2005; 4(6):987-95. doi: 10.1158/1535-7163.mct-05-0024 [Crossref] [ Google Scholar]
- Bacon NA, Larre I, Lawag AA, Merritt C, 2nd 2nd, Smith M, Rosolen M. Low dose HSP90 inhibition with AUY922 blunts rapid evolution of metastatic and drug resistant phenotypes induced by TGF-β and paclitaxel in A549 cells. Biomed Pharmacother 2020; 129:110434. doi: 10.1016/j.biopha.2020.110434 [Crossref] [ Google Scholar]
- Han ML, Zhao YF, Tan CH, Xiong YJ, Wang WJ, Wu F. Cathepsin L upregulation-induced EMT phenotype is associated with the acquisition of cisplatin or paclitaxel resistance in A549 cells. Acta Pharmacol Sin 2016; 37(12):1606-22. doi: 10.1038/aps.2016.93 [Crossref] [ Google Scholar]
- Liston DR, Davis M. Clinically relevant concentrations of anticancer drugs: a guide for nonclinical studies. Clin Cancer Res 2017; 23(14):3489-98. doi: 10.1158/1078-0432.ccr-16-3083 [Crossref] [ Google Scholar]
- Sui X, Zhang R, Liu S, Duan T, Zhai L, Zhang M. RSL3 drives ferroptosis through GPX4 inactivation and ROS production in colorectal cancer. Front Pharmacol 2018; 9:1371. doi: 10.3389/fphar.2018.01371 [Crossref] [ Google Scholar]
- Imai H, Matsuoka M, Kumagai T, Sakamoto T, Koumura T. Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis. Curr Top Microbiol Immunol 2017; 403:143-70. doi: 10.1007/82_2016_508 [Crossref] [ Google Scholar]
- Jiang M, Qiao M, Zhao C, Deng J, Li X, Zhou C. Targeting ferroptosis for cancer therapy: exploring novel strategies from its mechanisms and role in cancers. Transl Lung Cancer Res 2020; 9(4):1569-84. doi: 10.21037/tlcr-20-341 [Crossref] [ Google Scholar]
- Sun Y, Chen P, Zhai B, Zhang M, Xiang Y, Fang J. The emerging role of ferroptosis in inflammation. Biomed Pharmacother 2020; 127:110108. doi: 10.1016/j.biopha.2020.110108 [Crossref] [ Google Scholar]
- Bhattacharjee S. Reactive oxygen species (ROS): modulator of response to cancer therapy in non-small-cell lung carcinoma (NSCLC). In: Chakraborti S, Parinandi NL, Ghosh R, Ganguly NK, Chakraborti T, eds. Oxidative Stress in Lung Diseases. Vol 2. Singapore: Springer; 2020. p. 363-83. 10.1007/978-981-32-9366-3_16.
- Gill JG, Piskounova E, Morrison SJ. Cancer, oxidative stress, and metastasis. Cold Spring HarbSymp Quant Biol 2016; 81:163-75. doi: 10.1101/sqb.2016.81.030791 [Crossref] [ Google Scholar]
- Perillo B, Di Donato M, Pezone A, Di Zazzo E, Giovannelli P, Galasso G. ROS in cancer therapy: the bright side of the moon. Exp Mol Med 2020; 52(2):192-203. doi: 10.1038/s12276-020-0384-2 [Crossref] [ Google Scholar]
- Wang H, Zhang G, Zhang H, Zhang F, Zhou B, Ning F. Acquisition of epithelial-mesenchymal transition phenotype and cancer stem cell-like properties in cisplatin-resistant lung cancer cells through AKT/β-catenin/Snail signaling pathway. Eur J Pharmacol 2014; 723:156-66. doi: 10.1016/j.ejphar.2013.12.004 [Crossref] [ Google Scholar]
- Wang M, Liu ZM, Li XC, Yao YT, Yin ZX. Activation of ERK1/2 and Akt is associated with cisplatin resistance in human lung cancer cells. J Chemother 2013; 25(3):162-9. doi: 10.1179/1973947812y.0000000056 [Crossref] [ Google Scholar]
- Zhang Q, Zhang B, Sun L, Yan Q, Zhang Y, Zhang Z. Cisplatin resistance in lung cancer is mediated by MACC1 expression through PI3K/AKT signaling pathway activation. Acta BiochimBiophys Sin (Shanghai) 2018; 50(8):748-56. doi: 10.1093/abbs/gmy074 [Crossref] [ Google Scholar]
- Kim TR, Cho EW, Paik SG, Kim IG. Hypoxia-induced SM22α in A549 cells activates the IGF1R/PI3K/Akt pathway, conferring cellular resistance against chemo- and radiation therapy. FEBS Lett 2012; 586(4):303-9. doi: 10.1016/j.febslet.2011.12.036 [Crossref] [ Google Scholar]
- Zhang Y, Bao C, Mu Q, Chen J, Wang J, Mi Y. Reversal of cisplatin resistance by inhibiting PI3K/Akt signal pathway in human lung cancer cells. Neoplasma 2016; 63(3):362-70. doi: 10.4149/304_150806n433 [Crossref] [ Google Scholar]
- Qi Z, Wang Y, Zhou X. [CA916798 gene participates in cisplatin resistance of human lung adenocarcinoma A549 cells through PI3K/AKT/mTOR pathway]. Nan Fang Yi Ke Da XueXue Bao 2012; 32(9):1290-3. [ Google Scholar]
- Tang XL, Yan L, Zhu L, Jiao DM, Chen J, Chen QY. Salvianolic acid A reverses cisplatin resistance in lung cancer A549 cells by targeting c-met and attenuating Akt/mTOR pathway. J Pharmacol Sci 2017; 135(1):1-7. doi: 10.1016/j.jphs.2017.06.006 [Crossref] [ Google Scholar]
- Zuo Y, Yang D, Yu Y, Xiang M, Li H, Yang J. Niclosamide enhances the cytotoxic effect of cisplatin in cisplatin-resistant human lung cancer cells via suppression of lung resistance-related protein and c-myc. Mol Med Rep 2018; 17(3):3497-502. doi: 10.3892/mmr.2017.8301 [Crossref] [ Google Scholar]
- Wang M, Liu ZM, Li XC, Yao YT, Yin ZX. Activation of ERK1/2 and Akt is associated with cisplatin resistance in human lung cancer cells. J Chemother 2013; 25(3):162-9. doi: 10.1179/1973947812y.0000000056 [Crossref] [ Google Scholar]
- Zhou YT, Li K, Tian H. Effects of vinorelbine on cisplatin resistance reversal in human lung cancer A549/DDP cells. Asian Pac J Cancer Prev 2013; 14(8):4635-9. doi: 10.7314/apjcp.2013.14.8.4635 [Crossref] [ Google Scholar]
- Zhang K, Wang X, Wang H. Effect and mechanism of Src tyrosine kinase inhibitor sunitinib on the drug-resistance reversal of human A549/DDP cisplatin-resistant lung cancer cell line. Mol Med Rep 2014; 10(4):2065-72. doi: 10.3892/mmr.2014.2440 [Crossref] [ Google Scholar]
- Yu M, Qi B, Xiaoxiang W, Xu J, Liu X. Baicalein increases cisplatin sensitivity of A549 lung adenocarcinoma cells via PI3K/Akt/NF-κB pathway. Biomed Pharmacother 2017; 90:677-85. doi: 10.1016/j.biopha.2017.04.001 [Crossref] [ Google Scholar]
- Han L, Cao X, Chen Z, Guo X, Yang L, Zhou Y. Overcoming cisplatin resistance by targeting the MTDH-PTEN interaction in ovarian cancer with sera derived from rats exposed to Guizhi Fuling wan extract. BMC Complement Med Ther 2020; 20(1):57. doi: 10.1186/s12906-020-2825-9 [Crossref] [ Google Scholar]
- Persaud L, De Jesus D, Brannigan O, Richiez-Paredes M, Huaman J, Alvarado G. Mechanism of action and applications of interleukin 24 in immunotherapy. Int J Mol Sci 2016; 17(6):869. doi: 10.3390/ijms17060869 [Crossref] [ Google Scholar]
- Panneerselvam J, Munshi A, Ramesh R. Molecular targets and signaling pathways regulated by interleukin (IL)-24 in mediating its antitumor activities. J Mol Signal 2013; 8(1):15. doi: 10.1186/1750-2187-8-15 [Crossref] [ Google Scholar]
- Wei Y, Wu S, Xu W, Liang Y, Li Y, Zhao W. Depleted aldehyde dehydrogenase 1A1 (ALDH1A1) reverses cisplatin resistance of human lung adenocarcinoma cell A549/DDP. Thorac Cancer 2017; 8(1):26-32. doi: 10.1111/1759-7714.12400 [Crossref] [ Google Scholar]
- He R, Liu H. TRIM59 knockdown blocks cisplatin resistance in A549/DDP cells through regulating PTEN/AKT/HK2. Gene 2020; 747:144553. doi: 10.1016/j.gene.2020.144553 [Crossref] [ Google Scholar]
- Li A, Song J, Lai Q, Liu B, Wang H, Xu Y. Hypermethylation of ATP-binding cassette B1 (ABCB1) multidrug resistance 1 (MDR1) is associated with cisplatin resistance in the A549 lung adenocarcinoma cell line. Int J Exp Pathol 2016; 97(6):412-21. doi: 10.1111/iep.12212 [Crossref] [ Google Scholar]
- Abdalkader M, Lampinen R, Kanninen KM, Malm TM, Liddell JR. Targeting Nrf2 to suppress ferroptosis and mitochondrial dysfunction in neurodegeneration. Front Neurosci 2018; 12:466. doi: 10.3389/fnins.2018.00466 [Crossref] [ Google Scholar]
- Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi AA. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther 2021; 6(1):49. doi: 10.1038/s41392-020-00428-9 [Crossref] [ Google Scholar]
- Zuo S, Yu J, Pan H, Lu L. Novel insights on targeting ferroptosis in cancer therapy. Biomark Res 2020; 8:50. doi: 10.1186/s40364-020-00229-w [Crossref] [ Google Scholar]
- Kuang F, Liu J, Tang D, Kang R. Oxidative damage and antioxidant defense in ferroptosis. Front Cell Dev Biol 2020; 8:586578. doi: 10.3389/fcell.2020.586578 [Crossref] [ Google Scholar]
- Kajarabille N, Latunde-Dada GO. Programmed cell-death by ferroptosis: antioxidants as mitigators. Int J Mol Sci 2019; 20(19):4968. doi: 10.3390/ijms20194968 [Crossref] [ Google Scholar]
- Roh JL, Kim EH, Jang H, Shin D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol 2017; 11:254-62. doi: 10.1016/j.redox.2016.12.010 [Crossref] [ Google Scholar]
- Kwon OS, Kwon EJ, Kong HJ, Choi JY, Kim YJ, Lee EW. Systematic identification of a nuclear receptor-enriched predictive signature for erastin-induced ferroptosis. Redox Biol 2020; 37:101719. doi: 10.1016/j.redox.2020.101719 [Crossref] [ Google Scholar]
- Lee MW, Kim DS, Min NY, Kim HT. Akt1 inhibition by RNA interference sensitizes human non-small cell lung cancer cells to cisplatin. Int J Cancer 2008; 122(10):2380-4. doi: 10.1002/ijc.23371 [Crossref] [ Google Scholar]
- Han Z, Hong L, Wu K, Han S, Shen H, Liu C. Reversal of multidrug resistance of gastric cancer cells by downregulation of Akt1 with Akt1 siRNA. J Exp Clin Cancer Res 2006; 25(4):601-6. [ Google Scholar]
- Khwaja A. Akt is more than just a Bad kinase. Nature 1999; 401(6748):33-4. doi: 10.1038/43354 [Crossref] [ Google Scholar]