Advanced pharmaceutical bulletin. 14(1):48-66.
doi: 10.34172/apb.2024.016
Review Article
Lipid-Based Nanoparticles as Oral Drug Delivery Systems: Overcoming Poor Gastrointestinal Absorption and Enhancing Bioavailability of Peptide and Protein Therapeutics
Soheil Mehrdadi * 
Author information:
Department of Pharmaceutical and Pharmacological Sciences, University of Padova, Padua, Italy.
Abstract
Delivery and formulation of oral peptide and protein therapeutics have always been a challenge for the pharmaceutical industry. The oral bioavailability of peptide and protein therapeutics mainly relies on their gastrointestinal solubility and permeability which are affected by their poor membrane penetration, high molecular weight and proteolytic (chemical and enzymatic) degradation resulting in limited delivery and therapeutic efficacy. The present review article highlights the challenges and limitations of oral delivery of peptide and protein therapeutics focusing on the application, potential and importance of solid lipid nanoparticles (SLNs) and nanostructured lipid carriers (NLCs) as lipid-based drug delivery systems (LBDDSs) and their advantages and drawbacks. LBDDSs, due to their lipid-based matrix can encapsulate both lipophilic and hydrophilic drugs, and by reducing the first-pass effect and avoiding proteolytic degradation offer improved drug stability, dissolution rate, absorption, bioavailability and controlled drug release. Furthermore, their small size, high surface area and surface modification increase their mucosal adhesion, tissue-targeted distribution, physiological function and half-life. Properties such as simple preparation, high-scale manufacturing, biodegradability, biocompatibility, prolonged half-life, lower toxicity, lower adverse effects, lipid-based structure, higher drug encapsulation rate and various drug release profile compared to other similar carrier systems makes LBDDSs a promising drug delivery system (DDS). Nevertheless, undesired physicochemical features of peptide and protein drug development and discovery such as plasma stability, membrane permeability and circulation half-life remain a serious challenge which should be addressed in future.
Keywords: Peptide and protein therapeutics, Lipid-based drug delivery systems, Solid lipid nanoparticles, Nanostructured lipid carriers, Oral drug delivery
Copyright and License Information
©2024 The Authors.
This is an Open Access article distributed under the terms of the Creative Commons Attribution (CC BY), which permits unrestricted use, distribution, and reproduction in any medium, as long as the original authors and source are cited. No permission is required from the authors or the publishers.
Introduction
In recent decades, high demands for cost-benefit healthcare expenses, efficient therapeutics (e.g. safety and efficacy) and non-stop generic substitution has urged pharmaceutical companies,1 industry and market to shift to biotechnology-driven peptide and protein therapeutics and biopharmaceuticals. These novel categories of drugs, unlike active pharmaceutical ingredients (APIs, drugs), offer better feedback due to higher potency, selectivity and specificity for their extracellular target.2
Peptides and proteins, as cell products, have various physiological functions in body such as hormones, enzyme substrates and inhibitors, antibiotics, biological regulators, structural components, signaling factors and catalyzers which all implies their importance in body; hence, any abnormality in their amino acid sequence or structural disfunction leads to sever diseases and pathological conditions; diabetes,3 dwarfism,4 cystic fibrosis,5 thalassemia6 or impaired blood clotting,7 among many others.8,9 as such, due to their biological specificity and efficient affinity and efficacy, peptides and proteins have been exploited as drugs for treatment of diseases (Figure 1).10
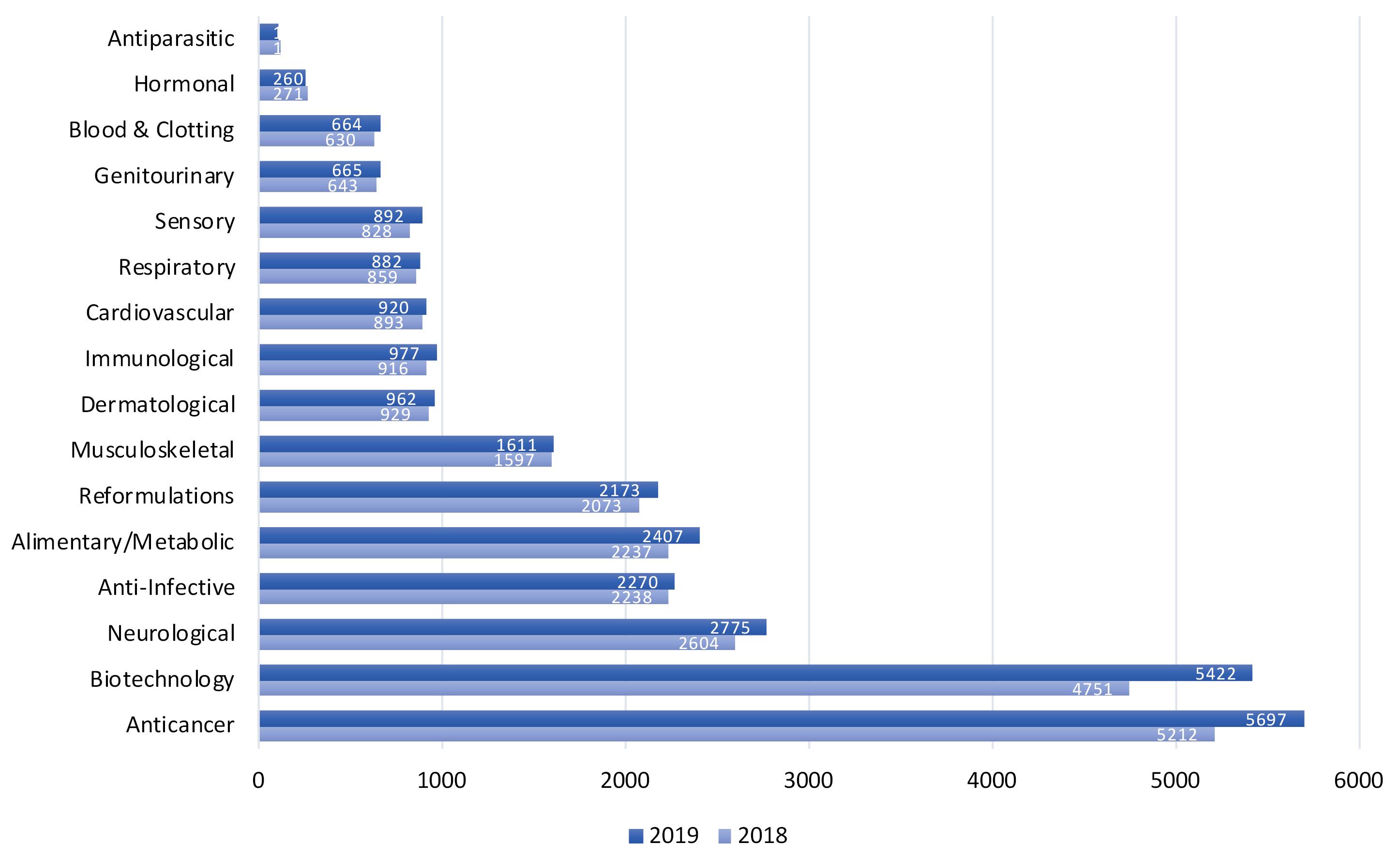
Figure 1.
Biotechnology medicines in research and development (R&D) by therapeutic category
.
Biotechnology medicines in research and development (R&D) by therapeutic category
Stability in proteins is the result of balance among four destabilizing and stabilizing forces: electrostatic interactions, hydrogen bonding, van der Waals forces and hydrophobic interactions, which form the secondary, tertiary and quaternary structures of proteins and any disruption will influence the structural balance and destabilizes that particular protein.11,12 Different environmental factors can influence the chemical and physical stability of proteins such as pH, ionic strength, temperature, high pressure, non-aqueous solvents, metal ions, detergents, adsorption, agitation and shearing which all are inevitably part of the manufacturing, sterilization and lyophilization process, and consequently might damage the developing protein resulting in biological inactivation, aggregation, immunogenicity and precipitation.13-15
Peptide and protein therapeutics are highly dependent on the production process, yet are biocompatible, cost-benefit, with modifiable in-vivo bioactivity, specific targeting, chemical diversity, and easily synthesized by using solid-phase peptide synthesis methodologies (e.g. Merrifield’s method) in which the amino acid sequence can be precisely chosen and inserted at the molecular level by modifying the basic units.16 nevertheless, undesired physicochemical features of protein drug development and discovery, remains a serious challenge for formulation scientists as well as pharmaceutical and biotechnology companies.
Only recently peptides have been considered as therapeutic agents while they were never considered as a potential therapeutic agent17; mostly due to their protease degradation, metabolic instability, short half-life, manufacturing complications and high expenses, which in long-term administration renders them unfavorable in terms of patient costs and compliance especially with regard to parenteral administration as the majority of peptides (10%) have a very low oral bioavailability.18 On the other hand, peptides’ biodegradability into non- to low-toxicity metabolites,2 low drug-drug interactions and immunogenicity,2 higher tissue penetration (owing to their small size), higher in-vivo activity (per unit mass), stability and lower expenses favors them over large therapeutic proteins and antibodies for regulatory approval (higher than 20%) which is twice the rate of small molecules.17
As well as peptides, proteins also gained importance and their application in pharmaceutical science and industry was emphasized due to more advanced analytical methods resulting in the recognition of various peptides and hormones as therapeutic biopharmaceuticals, novel genetic and molecular engineering methods to produce large-scale proteins and recently-defined roles of proteins as regulatory components of numerous diseases.19,20 Since then, pharmaceutical industry has developed various large-scale oral delivery technologies for peptides/proteins as active ingredients.21
Low oral bioavailability of peptide and protein therapeutics renders them being formulated as parenteral preparations; large molecular size,22 susceptibility to enzymatic degradation (local or mostly GI tract), poor stability in the gastric acidic environment,23 poor intestinal penetration, short plasma half-life, immunogenicity and the propensity to aggregation, adsorption, and denaturation.24,25 Enzymatic degradation and poor intestinal penetration, among all, have been mainly mentioned for low oral bioavailability and short half-life of protein drugs ( < 1%) which is claimed to be increased to 30%-50% by pharmaceutical enterprises.26,27 Pharmaceutical therapeutic proteins, due to their large molecular size which leads to low blood absorption and diffusion, requires specific epithelial transporters otherwise they cannot enter the general circulation by the ordinary routes of drugs ansorption. Furthermore, low pH and protease enzymes of GI aggravate this condition even more.28
Among different administration routes, the most common route for peptide and protein therapeutics is intravenous (I.V.) injections (Table 1), which is not favorable in terms of patient compliance, clearance varies from a few minutes to several days, and might result in undesired deposition and distribution which require repeated injections with higher therapeutic doses to achieve efficacy,29,30 subsequently causing severe adverse effects.
Table 1.
Various peptides and proteins’ administration routes
|
Method
|
Delivery routes
|
Formulation/Device requirement
|
Reference
|
| Invasive |
Intravenous (I.V.), subcutaneous (S.C.), intramuscular (I.M.),
Intracerebral vein (I.C.V.) |
Liquid or reconstituted solid (syringe), i.v. injected liposomes |
31-35
|
| Depot system (S.C. or I.M.) |
Biodegradable polymers, liposomes, permeable polymers (not degradable) microspheres, implants |
31-33,36,37
|
| Non-invasive |
Pulmonary |
Liquid or powder formulations, nebulizers, metered dose inhalers, dry powder inhalers |
31-34,38
|
| Oral |
Solids, emulsions, microparticles, nanoparticles, with or without absorption enhancers |
31-33,38,39
|
| Nasal |
Liquid, usually requires permeation enhancers, nanoparticles |
31-33,40,41
|
| Transdermal |
Iontophoresis, electroporation, chemical permeation enhancers, prodrugs, sonophoresis, transfersomes |
31-33,42,43
|
| Buccal, rectal, vaginal, ocular |
Gels, suppositories, bioadhesives, microparticles |
31-33,44,45
|
The subcutaneous (S.C.) and intramuscular (I.M.) injections are other administration routes (Table 1), the former is the most common one, especially for vaccines. Different factors such as molecular weight, site of injection, local injection site activity and pathological conditions can influence the fate of peptide and protein therapeutics following S.C. injection leading to a bioavailability as high as 100% or much lower.46 upon injection, proteins with high molecular weight ( < 16 000 Da) are absorbed either from vessels’ endothelial cells to capillaries or reach the local lymphatic system and through thoracic duct join the blood circulation, while proteins with small molecular weight are mainly diffused through the local capillaries.46 however, protein transportation through lymphatic way is undesired due to slow time of circulation which might result in protein enzymatic degradation.46,47
Non-invasive routes have increasingly been investigated as well as an alternative to the conventional invasive injectable routes (Table 1). Numerous studies have investigated nasal, ophthalmic, buccal, rectal, vaginal, transdermal and pulmonary routes for peptide and protein delivery.28,48-55
Based on studies, mucosae which so far have been neglected for drug delivery seem to be a promising approach for drug absorption, especially efficient for biomolecules of large size and molecular weight.28,56 The advantages of mucosal surfaces (mouth, eye, nose, rectum and vagina) for drug delivery over skin and GI tract can be named as: fewer biological barriers to pass for systemic diffusion, rapid absorption and evading hepatic first-pass effect. However, one practical challenge of mucosae is related to the preparations that are formulated for local long-term treatment.
Despite alternate routes of drug delivery, oral delivery (P.O.) is still the most preferred one; non-invasive, painlessness, easy self-administration, low risk of cross-infection, high patient convenience/compliance, outpatient feasibility,57 cost-benefit (no need for sterile manufacturing).57 Oral route does not offer the drawbacks of I.V. route; drug extravasation from blood, catheter-related infectious complications, thrombosis and being expensive and invasive, especially for chronic conditions.
However, biological barriers of GI tract with their associated enzymatic and chemical processes hamper the efficiency of oral route for drug delivery. Furthermore, the epithelial cell monolayer membrane of the GI tract even more aggravate the condition of low permeability for many peptide and protein therapeutics with low gastrointestinal solubility which finally results in low bioavailability.58
Some active moieties cannot be delivered through oral route.59,60 According to the Biopharmaceutic Classification System (BCS),61 oral bioavailability of each drug is determined by its solubility along the GI tract and cellular penetration. Most of the potential drug candidates developed with high-throughput screening methods generally have higher molecular weights and tend to be lipophilic in nature.62 Other factors contributing to low oral bioavailability of drugs are low stability in the gastrointestinal environment and poor membrane permeability. Most of drugs are substrates to intestinal efflux transporters like p-glycoprotein resulting in poor oral bioavailability.63
Regardless of the administration route, majority of peptide and protein therapeutics, lacking necessary physicochemical requirements, fail to diffuse and be absorbed to their target tissue which implies the importance of drug delivery and tissue-targeting systems for achieving as maximum therapeutic effects as possible. The carrier systems diffuse and distribute the intended therapeutic molecules to their targeted site with as maximum concentration as possible in the affected area and as minimum concentration as possible in the intact tissues to lower the general adverse effects.64
Peptide and Protein Drug Delivery
Introduction of novel biotechnological molecules as potential therapeutics, advent of chemical synthesis methods and recombinant DNA technology have all rendered protein synthesis and delivery an important area of research which resulted in the production of numerous large-scales drugs of peptide/protein origin such as monoclonal antibodies, hormones and vaccines. According to The 2018 and 2019 PhRMA reports,65 there have been respectively 4751 and 5422 novel biotechnological medicines in research and development (R&D) phase for more than 100 diseases such as cancer, infectious diseases, autoimmune diseases, AIDS/HIV, antiparasitic and related conditions (Figure 1), which have been either in human clinical trials or under review by the Food and Drug Administration (FDA).65 nevertheless, abovementioned challenges for their delivery through GI tract 66-70 and the blood brain barrier (BBB)(in the case of central nervous system diseases),71,72 makes their therapeutic potential and clinical application questionable.
In the last decades, numerous drugs of peptide/protein origin have been in preclinical studies and clinical trials,73 more than 400 recombinant peptides and proteins and 1300 under clinical trials.74 The reason could be attributed mostly to the larger size of peptides and proteins comparing to conventional drugs, which provides drug-target interaction with binding pockets that are not normally available to small molecular drugs. These targets could be part of intracellular protein-protein interaction network which have been recognized in numerous diseases. Peptide and protein therapeutics in order to interact with such targets must penetrate cells, however, most of them are known to have extracellular targets,73 and are parenterally administered so cellular penetration is not their ordinary route as it is for mucosal surfaces. Currently, the main obstacle of the oral administration of these novel categories of drugs for their maximum therapeutic effects could be addressed as the penetration through intestinal cellular membranes and target cellular membranes.
Transport mechanisms in the GI tract
To formulate and synthesize drug delivery systems (DDSs) for oral peptide and protein therapeutics, and biopharmaceuticals a throughput understanding of the biological pathways involved for their absorption and diffusion in the GI tract is necessary and worthwhile. Various physicochemical features govern the pathway through which the molecules will be penetrating the intestinal cells; molecular weight, hydrophobicity/hydrophilicity, ionization constants, and pH stability, among all.
Paracellular transport
It has the following features as, space dimension of 10 Å, aqueous pores (epithelial tight junctions) 7–9, 3–4 and 8–9 Å for the jejunum, the ileum and the colon respectively,75 to allow the passage of solutes with a specific molecular radius and tight junctions building 0.01% of the total absorption surface area of the intestine.76 These data prove the restriction of the paracellular transport toward the passing molecules (Table 2). however, there is an electrical resistance diversity and consequently ionic selectivity. In the latter case, also transcellular pathway’s collaboration adjusts rate and selectivity of export of ions and solutes and overall tissue-specific transport. The tight junction along with ion channels are involved in size and charge selectivity, ion concentration-dependent penetration, competitive-based penetration among different molecules, unordinary mole-fraction effects and pH-sensitivity.77 hydrogen bonding capacity and lipophilicity do not influence much the paracellular pathway.
Table 2.
Transport mechanisms in the GI tract
|
Transport Mechanisms
|
Process
|
Feature of Molecules
|
Note
|
| Paracellular |
Passive diffusion in intercellular spaces between epithelial cells, tissue-specific transport78,79 |
Ions, large substances and solutes < 15 Å (3.5 kDa)80 |
Restricted protein delivery due to tight junctions81 |
| Transcellular |
Intestinal transcytosis, Enterocytes and M cells82 |
Various physicochemical properties |
Limited transport of relatively low molecular-weight lipophilic drugs |
| Carrier-mediated |
Across the cell membrane or entire cell83 |
Utilized by small hydrophilic molecules 84 |
Small di-/tripeptides monosaccharides, and amino acids are transported transcellularly85 |
| Receptor-mediated |
Receptor specific ligand,86 endocytosis87 |
The physicochemical and metabolic features |
Direct delivery of hydrophilic ligands to liver, direct delivery of lipophilic ligands to the vena cava88 |
Transcellular transport
It is an endocytic process at apical membrane and the absorbed molecules are released at the basolateral membrane, glucose is also transported with this mechanism (Table 2). The protein-lipid ratio is very insignificant in the basolateral membrane due to its thinner and more permeable structure than the apical membrane. This transport mechanism is governed by various factors: different physicochemical properties of molecules, size, lipophilicity/hydrophobicity, hydrogen bond formation, surface charge, superficial ligands; the physiological condition of the GI system and the animal models studied for transport mechanism.89,90 There are mainly two types of primary intestinal epithelial cells for molecules transportation; Enterocytes and M cells, the former lining about 99% of the GI tract and the latter mainly the area of Peyer’s patches and the human follicle-associated epithelium (FAE) (antigen-specialized).91 M cells function as presenting and transporting peptides and proteins to the local lymphoid tissues for immune reactions and a vulnerable and available way for pathogenic organisms.92 Due to their great endocytosis and transcytosis capacity for transporting diverse molecules and biomaterials (e.g. nanoparticles),93,94 M cells could be used for oral delivery of peptides and proteins and finally through phagocytosis, adsorptive (through clathrin-coated pits and vesicles) and fluid phase endocytosis they adsorb macromolecules and microorganisms.95 Some studies demonstrated nanoparticles transportation through intestinal villi and contradicting the recent debates over the rate of particle absorption.96,97 There is a consensus on the transportation of the majority of particles in FAE,96,98,99 for which there have been studies on the Peyer’s patches and M cells involvement on various biomaterials absorbency. The transcellular mechanism however is not a desirable route for low molecular-weight lipophilic drugs. Overall, absorption by this mechanism is reduced in a great extent in the colon part of the large intestines in comparison to the paracellular mechanisms.100
Carrier-mediated transport
It is an active and energy-dependent transportation of specific molecules against their concentration gradient through specific membrane receptors, such as β-lactam antibiotics and angiotensin converting enzyme inhibitors, monosaccharides and amino acids (Table 2). In one study using Caco-2 cell monolayers, it was proved that the conjugated insulin is transported 5 to 15 times more through the transferrin receptor than then insulin receptor itself.101
Receptor-mediated transport
It has been investigated to evaluate the oral bioavailability of peptide and protein drugs by modifying receptor specific ligands-drug interaction. This mechanism has functions in different processes such as endocytosis (clathrin-mediated), phagocytosis, pinocytosis and potocytosis (nonclathrin-mediated) (Table 2). The absorption starts with the binding of molecules to their specific receptors and their internalization into endosomes with low acidic pH which might dissociate receptor-ligand bound and accordingly degrades endosomes. The absorbed peptide and protein access into systemic blood circulation with two distinct pathways: hepatic portal vein and intestinal lymphatic vessels, the amount of peptides and proteins absorbed through either of these two pathways depends greatly on the physicochemical features of the formulation. portal vein is the main pathway for the majority of orally administered peptide and protein drugs and through which hydrophilic molecules are absorbed and transported to the blood systemic circulation first through the hepatic portal vein and then by the hepatic artery, and finally they are delivered to their sites of action. But lipophilic molecules penetrating through the same intestinal barriers are transported to the intestinal lymphatic vessels, bypassing the first-pass effect, and directly delivered to the vena cava for blood systemic circulation.
Absorption of oral drugs
Drugs in order to be absorbed through GI tract are required to have high solubility and permeability, however, this is not the case for numerous drugs with low aqueous solubility and consequently low and diverse bioavailability.102 for such drugs simultaneous presence of high amount of fat through meals can increase their oral bioavailability,103-105 via prolonging GI tract passage time, exocrine pancreas secretion stimulation, reduced metabolism, lymphatic-associated absorption, increased intestinal penetration, reduced cellular efflux and liver- and mesenteric-related blood alteration.106,107
Introduction of lipid-based drug delivery systems (LBDDSs) in 1990s provided scaffolds which increase dissolution rate of poor aqueous soluble drugs (i.e. hydrophobic drugs) by providing a phase in which the drugs can disintegrate and be absorbed and diffused toward its site of action.108 after the degradation of lipids in the intestine, active mono- and diglycerides are formed on the surface of lipids which later disassociate and transform into micelles and simultaneously drug is also solubilized inside micelles. These micelle-drug mixtures are finally absorbed.109-112
The absorbed components through intestinal segment of GI system follow two distinct pathways according to their features: blood vessels and lymphatic vessels. The former is the preferred route for most of the oral drugs by which they are absorbed into the systemic circulation via portal vain, and the latter for highly lipophilic drugs (log P > 5) by which drugs are absorbed into systemic circulation via lymphatic vessels.
Overall, the presence of lipid increases absorption of numerous drugs more through lymphatic vessels, especially lipophilic ones and macromolecules of high molecular weight,113,114 mostly due to their higher permeability to nanoparticles than blood vessels,115 and most importantly overpassing hepatic first-pass effect.116 Nevertheless, it has been demonstrated that the absorption through lymphatic pathway is affected by the length of fatty acid chains; long-chain triglycerides (14-18 chains) are more favored for absorption than low-chain ones.111,117
Physiological barriers against peptide and protein therapeutics absorption
Gastrointestinal barriers
In-depth studies of molecular pharmacology have provided a better understanding of the biological and molecular processes of the GI system, while its target sites has introduced new insights for targeted delivery of oral peptide and protein therapeutics and biopaharmaceuticals.118,119 There are two types of proteolytic enzymes with their specific site of action which are responsible partly for the physiological processes of GI system toward peptides and proteins: endoproteases, including trypsin, chymotrypsin, and elastase, which hydrolyze the internal bonds of the peptide chain to the amino- and carboxy-terminus, and exopeptidases, including carboxypeptidase A and aminopeptidase, which hydrolyze the amino- and carboxy-terminus bonds to the peptide chain (Table 3).120 Enzymatic degradation happens at the lumen, brush border, the cytosol of the enterocytes, in the lysosomes and other cell organelles.121
Table 3.
Proteases and their sites of action
|
Types
|
Enzymes
|
Major Site of Action
|
| Gastric proteases |
Pepsins (aspartic proteases) |
Broad activity, hydrolyzes numerous peptide bonds |
| Brush border proteases |
Aminopeptidase A
Aminopeptidase N
Aminooligopeptidase
Dipeptidylaminopeptidase IV
Carboxypeptidase |
Aminopeptidases are N-terminopeptidases, degrading mostly 3–10 amino acid residue-dipeptides and amino acids |
| Cytosolic proteases |
Di- and tripeptidase |
2-3 Aminopeptide amino acids |
| Intestinal pancreatic proteases |
Trypsin (endopeptidase)
α-chymotrypsin (endopeptidase)
Elastase (endopeptidase)
Carboxypeptidases (exopeptidase) |
Peptide bonds of basic amino acids/peptides Peptide bonds of hydrophobic amino acids/peptides Peptide bonds of smaller and nonaromatic amino acids/peptides
A: C-terminal amino acid
B: C-terminal basic amino acid |
| Brush border proteases |
Aminopeptidase A
Aminopeptidase N
Amino oligopeptidase
Dipeptidyl aminopeptidase IV
Carboxypeptidase |
Aminopeptidases are N-terminopeptidases, degrading
mostly 3–10 amino acid residue-dipeptides and amino acids |
The secretions of stomach (hydrochloric acid, potassium chloride and sodium chloride) provide an acidic pH of 1.5-3.5 for the proteolysis of peptides and proteins by breaking them down into amino acids, dipeptides and tripeptides for absorption. The digestion of peptides and proteins starts with pepsin in the acidic environment of stomach (pH 2), however, the alkaline environment of the intestines (pH 6) inactivates pepsin. The instantaneous wide change of pH from acidic stomach to alkaline intestines influences the degradation of ingested peptides and proteins and might contribute to their precipitation that redissolve following pH change.122-124
The small intestine is the major site of absorption along the GI tract due to the higher enzymatic activity of proteases (mostly in duodenum and jejunum). The brush borders of the intestinal epithelial cells secrete numerous specific enzymes (e.g. sucrose) leading to peptide/protein absorption and degradation.120 furthermore, the digestive secretions of the exocrine part of the pancreas also contain endo-/exopeptidases which are released into duodenum to increase pH for the activity of intestinal enzymes (e.g. trypsin). However, in the terminal parts of jejunum and ileum the enzymatic activity of aminopeptidases is decreased to 20–30% where Peyer’s patches are located and these areas could be a potential site for peptide/protein drug delivery.98,125
Mucosal barriers
Mucus has a major importance and function by determining the absorption and bioavailability of administered drugs especially through oral route. The mucosal surface of stomach has three components which further hamper the drug diffusion and absorption as an exogenous component: the first one, which is lined by surface epithelial cells and tight junctions, has a role against irritant and unsuitable fluids; the second one has a very unique insoluble protective mucus (the mixture of surface epithelial cells and neck cell) which creates a jelly-like layer throughout the entire surface mucosae of the stomach; and the third one is composed of bicarbonate ions which are secreted by the surface epithelial cells.126,127
There are however other components which further disturb peptide/protein absorption through oral administration, such as glycocalyx located on the surface layer of the stomach epithelial cells with an acidic nature and containing sulfated mucopolysaccharides. Goblet cells of the stomach wall secrete mucus, which covers upper layer of glycocalyx,128 and contains mucin glycoproteins, enzymes, electrolytes and water.129 the mucin glycoprotein gives glycocalyx an adhesive feature and functions more as a physical barrier than a chemical one,130,131 and in the stomach and colon parts of GI tract has a thick layer while in the small intestine part is thinner and this fact can be justified according to the digestive functions of each segment.132 Overall, the abovementioned components altogether are protected by viscoelastic layers. Peptide and proteins first of all are required to pass the outermost layers, mucus and glycocalyx, to reach the cellular membranes which present a viscous barrier to absorption and diffusion.
Nanomedicines – novel drug delivery systems
After administrating drugs, their plasma concentration increases and they affect their site of action effectively followed by a gradual decrease to a point when their plasma concentration is not sufficient enough anymore to elicit their intended therapeutic effect. Hence, Drugs are required to be re-administrated based on the dose and frequency of administration to provide the same concentration which must be neither higher nor lower than their therapeutic concentration level; a concept widely known as “therapeutic window”; higher doses will result in general toxic effects and lower ones cannot elicit any therapeutic effects.133 the conventional drugs do not have prolonged drug delivery features in their “therapeutic window” and this issue implies the importance of novel DDSs to be introduced.
A novel interdisciplinary branch of biomedical science, “nanomedicine”, has been investigating the potential application of biomaterials and nanostructures as DDSs for sustained, controlled and tissue-targeting delivery of drugs and active agents, which due to their pharmaceutical features overcome some of the limitations of conventional drugs.
Studies have proved that “drug discovery” alone does not offer practical solutions to therapeutics as most of the successful in-vitro experiments result in failure in in-vivo experiments mostly due to: poor drug concentration owing to low absorption, quick metabolism and elimination (peptides and proteins); general blood distribution which results in drug-related adverse effects and toxicity (anti-cancer drugs); low drug solubility of aqueous solutions when administered intravenously; unpredictable bioavailability and high plasma-level variations with oral administration and physiological processes involving a drug’s plasma level (e.g. food on cyclosporine).
In DDSs the in-vivo destiny of drugs is directly influenced by a series of factors which can be modified for in-site delivery with the desired therapeutic concentration. Peptide and protein therapeutics are encapsulated in nanomedicines for transportation through GI tract offering benefits such as high stability for storage and administration, and large-scale sterile manufacturing for oral preparations.134 Nanomedicines can be formulated and modified with the desired criteria such as size, surface properties and release profile to have tissue-targeted delivery within drug’s unique therapeutic window.
Nanomedicines have a size ranging from 1 to 100 nm where the therapeutic molecules can be incorporated in the core, matrix or attached on the surface (in the case of high surface/volume ratio),135 which the latter results in longer half-life and systemic circulation and increased mean residence time (MRT).136 Since 1990s LBDDSs have been under investigation owing to their advantages; biocompatibility, higher penetration capacity, lipophilicity with no need for surface modification, simple fabrication, cost benefit and industrial-scale production compared to their counterpart DDSs such as polymeric and inorganic nanomedicines.137,138
Based on the fabrication methods and physicochemical properties LBDDSs are classified into the following:
Liposomes in the form of spherical vesicles which are composed of one or multiple lipid bilayer (phospholipid or natural phospholipids) enclosing an aqueous core.137,138 Their size varies from 10 to 1000 nm and they are the first generation of LBDDSs that were employed mainly for parenteral route of drug delivery. They possess low antigenicity and toxicity, high drug encapsulation and loading efficiency and sustained and controlled drug release as their benefits. However, their synthesis is complex and has low stability and rapid reticuloendothelial system clearance and scale-up problems. Despite introducing various surface functionalization (antibodies and peptides) and modification (e.g. PEG coating) which enhances their blood half-time, structural stability and therapeutic efficiency, they are still undesirable in terms of industrial scale production.139-141 Currently, numerous liposome-based formulations are approved for a variety of diseases: Doxorubicin for cancers (Doxil®, Myocet® and Lipodox®), Amphotericin B for fungal infections (Ambisome®), Cytarabine for lymphomatous meningitis (Depocyt®), Morphine sulfate for pain management (DepoDur®), inactivated hepatitis A virus (strain RG-SB) for hepatitis A (Epaxal®) and inactivated hemagglutinin of Influenza virus strains A and B for influenza (Inflexal®).142
Lipoplexes are fabricated from liposomes and are designed in multi-lamellar lipoplexes with positive lipid bilayer and distinct negative nucleic acids. The electrostatic process between self-assembly liposomes and nucleic acids produces such scaffolds. Given their similarity to liposomes they possess similar benefits and drawbacks with a few of their own; multiple cations tendency to bind negative nucleic acids which decreases the internal cell transfection process. They have been investigated for brain-focused studies.143
Natural body lipoproteins are another lipid-based system which are very similar to liposomes hence sharing the same advantages and disadvantages and carry lipids (mainly cholesterol), proteins, enzymes, and miRNAs. They have been investigated with other nanoparticles (e.g., albumin, PEG-PLGA) for numerous central nervous system diseases.144
(2) Niosomes are vesicles with a lamellar self-assembled structure which are composed of non-ionic surfactants and cholesterol or its derivatives.145 They can be encapsulated by lipophilic and hydrophilic substances. They are cheaper in terms of production and more stable than liposomes.146
(3) Transferosomes which are composed of a lipid bilayer fabricated by a lipid matrix stabilized by various surfactants,147 and are similar to niosomes and to liposomes.
(4) Solid lipid nanoparticles (SLNs) which are composed of a solid lipid core.148
(5) Nanostructured lipid carriers (NLCs) which are composed of a liquid lipid phase core within the solid lipid phase.149
Among all the lipid-based DDSs, this review will focus further on SLNs and NLCs and their unique characteristics and properties. SLNs, comparing to NLCs which are formulated with both solid and liquid oils, are formulated only with solid ones which gives them more controlled drug release due to limited drug mobility in solid lipids and are designed as oral pellets and retard capsule (e.g. Mucosolvan®), as microparticles by spray drying,150 and oral nanopellets.151
Solid lipid nanoparticles and nanostructured lipid carriers
SLNs and NLCs are lipid-based colloidal drug carriers, synthesized from lipids (solid or liquid), surfactants, co-surfactants and APIs (drugs). SLNs are composed of solid lipids and surfactants but NLCs are also composed of liquid lipids and oils. Lipids have solid form both at room and body temperature and could be chosen as purified triglycerides, glyceride mixtures and waxes. Surface surfactants increase and improve the stability and cellular permeability and consequently absorption.152,153
They collectively offer the benefits of other colloidal DDSs (e.g. liposomes and polymeric NPs) and avoid their drawbacks;154 enhanced dissolution rate, bioavailability, tissue distribution, encapsulation rate, absorption, stability of drug in body fluids, no unpleasant taste (exists with oral preparations), lower toxicity, no organic solvents usage, large-scale production possibility, sterility, reduced first-pass effect and controlled and sustained tissue-targeted delivery with various routes of administration: oral, parenteral, nasal, rectal, ophthalmic, etc.155
Due to some drawbacks of SLNs such as low drug loading capacity, unpredictable gelation tendency, and drug expulsion after polymorphic transition during storage,152-156 NLCs were introduced and synthesized as the improved version of lipid-based nanocarriers with numerous methods exploited to formulate and prepare them.155-157 Hence, the final nanoparticle characterization must comply with the dynamic processes and such a characterization is the real challenge to represent the highest quality for the product,157 storage drug expulsion, unpredictable gelation and their co-encapsulation with nano- and micro-sized structures owing to high lipid concentration and surfactants might influence the in-vivo fate of nanoparticles.
The major criteria for the characterization of nanoparticles as efficient and safe DDSs could be addressed as: size, encapsulation efficiency, structure, co-existence of material, surface morphology and functionalization, and minimum drug dose, which arguably influence directly the bioavailability, absorption and distribution of the encapsulated drug (Table 4).
Table 4.
Models of drug incorporation for the lipid nanoparticles
|
Model
|
Drug Loading Site
|
Drug Release Pattern
|
| Homogenous matrix of solid solution158,159 |
Homogeneous drug dispersion in the lipid matrix of the particles |
Diffusion from the solid lipid matrix and/or by degradation of lipid matrix in GI |
| Drug-enriched shell158,159 |
Drug concentration on the outer shell of the nanoparticles |
Burst release160 modified by varying the formulation conditions: production temperature (preferably cold homogenization) and surfactant concentration161 |
| Drug-enriched core158,159 |
Drug concentration in the core of the nanoparticles |
Prolonged drug release161 |
Types of solid lipid nanoparticles
Type 1: Drug molecules/APIs are dispersed either in the lipid core or as amorphous clusters in “the homogenous matrix model”, which offers controlled release features. In order to design and fabricate this type, appropriate concentration of API/lipid ratio must be adjusted using either above-melting point of the lipid or cold methods of high-pressure homogenization (HPH).162
Type 2: This type, known as “Drug enriched shell model”, is designed and fabricated with low concentration of API in the melted lipid. Using the hot method of HPH, the lipid phase is precipitated during the cooling phase which leaves a higher concentration of API in the residue of melted lipid leading to the formation of a free-API lipid core being surrounded by an outer shell composed of the saturated API and lipid. This type does not function for sustained release, however, it does for burst release of API.162
Type 3: Hence “Drug enriched core model”, it is designed by solubilizing the drug in the melted lipid up to its saturation solubility. Following cooling of the lipid the drug is super-saturated in the melted lipid and recrystallizes before the lipid does. Further cooling process renders also lipid recrystallization surrounding the prior formed drug-enriched core. This type offers sustained and prolonged drug release.162
Types of nanostructured lipid carriers
Imperfect. They are named “imperfect” due to the tiny pores in the solid matrix core which are loaded with API. They are designed by adding and blending solid and liquid lipids (oil) which the co-presence of fatty acids with different chain length (mono-, di- and triacylglycerols) confers an imperfect structure for encapsulating the API.163
Amorphous. They are designed blending lipids which don’t crystallize after homogenization and cooling process,164 such as hydroxyl octacosanyl hydroxy stearate, isopropyl myristate and dibutyl adipate, that give them an unorganized amorphous matrix which reduces API repelling of storage and shelf time.163
Multiple. The advantage of the higher solubility of lipophilic drugs in liquid lipids than solid lipids can be used for formulating multiple types of NLC. Solid lipids are mixed with oils which are gradually added in higher amounts exceeding their solubility which results in phase separation of tiny particles with the surrounding solid lipid matrix. This type offers controlled drug release without expelling it out of the lipid matrix.163
Nevertheless, even the lipid-based DDSs have their own advantages and disadvantages (Table 5).
Table 5.
Advantages and disadvantages of lipid-based drug delivery systems
|
Characteristics
|
Biological/Technological aspects
|
Perspectives
|
Reference
|
|
|
Various administration routes |
Broad-spectrum drug application, therapy optimization |
165
|
|
|
Biodegradability |
Sustained drug release |
166
|
|
|
Controlled drug release |
Patients safety, prolonged drug release, in-site drug concentration |
167
|
|
|
Site-specific targeting |
Decreased systemic toxicity, targeted therapy |
168
|
|
|
Biocompatibility |
No allergic reactions |
169
|
|
|
Increased bioavailability of encapsulated drug |
Decreased dose |
170
|
|
|
Decreased adverse effects of toxic drugs |
Improved patient safety |
171
|
|
|
Biological barriers penetration |
Various administration routes |
172
|
|
|
Reduced dosing frequency |
Patients compliance |
167
|
| Advantages |
Chemical and enzymatic degradation drug protection |
Possibility of broad-spectrum administration routes |
168
|
|
|
Physical stability |
Improved drug formulation stability |
173
|
|
|
Capacity to encapsulate hydrophilic and hydrophobic drugs |
Versatility for different drug groups |
174
|
|
|
Scaled up production |
Industrial production possibility |
165
|
|
|
Simple manufacturing |
Easy fabrication in labs, low cost |
166
|
|
|
No organic solvents |
No toxicity concerns, green chemistry |
169
|
|
|
Co-delivery |
Offering combined therapy |
166
|
|
|
Increased drug loading capacity |
Decreased formulation dose |
173
|
|
|
Sterilization possibility |
Parenteral administration optimization |
175
|
|
|
Small size distribution |
Potential alternative for drug delivery |
176
|
|
|
Initial burst effect of encapsulated drug |
Patient overdose risk |
177
|
|
|
Low plasma circulation time |
Fast reticuloendothelial clearance before in-site deposition |
178
|
|
|
Drug expulsion during storage |
Storage and administration stability challenges, industrial-scale limitation |
179
|
| Disadvantages |
Low drug loading capacity |
High requirement of formulation doses |
180
|
|
|
Polydispersity |
Undesirable for intravenous administration |
181
|
|
|
Agglomeration |
Storage issues |
182
|
|
|
Storage in refrigerated conditions |
Transportation issues, expensive storage costs |
183
|
|
|
High operative temperature |
Susceptibility of thermolabile drugs |
184
|
Mechanisms of drug release by SLNs/NLCs
To improve the benefits and avoid the drawbacks, different criteria have been considered for formulation, design and encapsulation rate of nanomedicines.185 The encapsulated drug undergoes surface dissolution and degradation of the lipid matrix which results in diffusion of molecules from the matrix into the surrounding tissue.158 Drug release from SLNs/NLCs is affected by the localization of the drug,186 which can be loaded both in the core matrix and on the surface, the former results in prolonged and sustain release while the latter burst release (quick early-phase release), subsequently conferring a biphasic release profile starting with an quick release owing to the surface-loaded drug continuing by sustained release of more loaded-drug from the lipid matrix.
The burst release is a feature determined by modifying drug aqueous solubility which is influenced mainly by the surfactant concentration and the temperature via a direct proportion; the higher the last two the higher the burst release. Preparation of SLN/NLC nanoparticles at room temperature has demonstrated no burst release owing to no drug partitioning into water phase and following re-partitioning into lipid phase. Therefore, to decrease the burst release SLNs/NLCs have been prepared without surfactants or surfactants not solubilizing the drug.185,187
In vivo fate of SLNs/NLCs after administration
There are different criteria which determine the in vivo fate of nanoparticles: administration route, biological interactions with their environment including distribution, surface adsorption, nanoparticle disaggregation and enzymatic degradation.
Due to the presence of lipids and waxes in the structure of lipid-based nanoparticles, their fate is highly affected by different pathways and enzymes for their biological interactions, namely as lipases which exist ubiquitously in body and mostly activate by oil/water interface,188-190 and actively confer various degradation rates to nanoparticles due to their formulation material 191-194 the free fatty acids of degradation have been studied by enzymatic test,195 which demonstrated lesser degradation with long-chain fatty acids of triglycerides and surfactants contained in nanoparticles. Surfactants (e.g. poloxamer 407, poloxamer 188) function either to fasten or postpone the degradation process of nanoparticles, as different surfactants (e.g. cetyl palmitate)192 have different chain lengths, and this feature could be used in nanoparticle preparation to render a more controlled drug release profile.
So far, there has been few studies,196 approving whether the presence of food in stomach would affect nanoparticles’ in-vivo function or not, and this still remains a dilemma to be solved. In one animal study increased bioavailability and blood half-time was reported with oral administration of lipid nanodispersions,197 and in another study the increased absorption of nanoparticles into lymph through intraduodenal route was reported.198
In vivo toxicity evaluation of SLNs/NLCs
Along with their different therapeutic application as DDSs, SLNs and NLCs have been investigated for their in-vivo toxicity/safety profile. Due to their composition of lipids which are physiological components they are generally recognized as safe (GRAS) and better-tolerated nanoparticles than polymeric ones showing lower toxicity,199,200 as they are degraded by normal physiological pathways. Nevertheless, the type of lipids and surfactants (emulsifiers) used for their preparation might increase or decrease cell toxicity and even influence encapsulated drug toxicity.201,202 Therefore, the toxicity evaluation must include bulk materials, SLNs/NLCs, drug/API itself and drug-encapsulated nanoparticles to analyze thoroughly each component and materials contribution to toxicity. The excipients role for drug encapsulation must be assessed according to the route of administration.203
Different in-vitro tests, among all cell viability (MTT assay) and oxidative stress, have been exploited to assess the cytotoxicity of nanoparticles; the former functions as the color-change of tetrazolium which is the indication of cell death, and the latter demonstrates DNA damages, elevated amounts of reactive oxygen species, lipid peroxidases and alterations in oxidation/reduction glutathione reactions.204 MTT assay is the most common method used with different dyes such as Neutral red, Trypan blue,204-206 and there are in-vitro and in-vivo experiments on cells proving their low toxicity.207,208
SLNs and NLCs co-delivery strategies
Through numerous cancer-related studies, it has been proved that broad-spectrum anti-cancer agents will have many benefits in terms of efficacy over monotherapy. SLNs and NLCs could be a promising carrier for co-delivery of anti-cancer, therapeutic nucleic acids and antibiotics,209 as they are significantly able to enhance the in-vitro and in-vivo therapeutic efficacy of such drugs. Besides such advantage, co-encapsulation of different drugs in one LPDDS might decrease toxicity of the respective anti-cancer drugs and other adverse effects coming with them separately.210
Another alternative being offered by co-delivery is RNA interference,211 especially siRNAs have been exploited for cancer cells to silence the oncogenes expression.209 In this context, miRNAs have been investigated and proved to be efficient to regulate genes associated with tumorigenesis.210
In one study,209 cationic SLNs for co-delivery of paclitaxel and human myeloid cell leukemia (MCL1) specific siRNA have been investigated and the final result demonstrated enhanced in-vitro and in-vivo efficacy than administering them separately.
In another similar study,210 SLNs were encapsulated for co-delivery of the same active substance with miRNA-34a. The final result was significant in terms of eliminating lung cancer relapse mostly owing to the synergic efficacy and higher inhibition of specific receptors.
In another study,212 the efficacy of Paclitaxel and Verapamil co-loaded SLNs toward breast cancer were investigated to prove the efficiency of verapamil for inhibiting drug efflux transporters (e.g. p-glycoprotein) on multidrug resistance cancer cells. The study demonstrated higher expression downregulation of g-glycoproteins in the specific cancer cells, as well as higher cellular drug uptake and toxicity comparing to Paclitaxel and sole anti-cancer administration.
In another study on antibiotics,213 increased antibacterial activity of Vancomycin was demonstrated. Ion pairing with linoleic acid was exploited for co-delivery and significant effects against Staphylococcus aureus infections which could be interpreted owing to the increased lipophilicity, sustained release of antibiotic and synergistic effect.
So far different pharmaceutical/biotechnological products have been marketed using lipid-based DDSs (Table 6).
Table 6.
List of marketed lipid-based oral pharmaceutical products
|
Product/Trade name
|
Drug/Molecule
|
Nanotechnology/Dosage form
|
Therapeutic use/Indication
|
Company/Alliance
|
| Abelcet |
Amphotericin B |
Nanoliposome/solution |
Fungal infections |
The Liposome Company Inc |
| Accutane |
Isotretinoin |
Emulsion/soft gelatine capsule |
Anti-comedogenic |
Roche |
| Agenerases |
Amprenavir |
Soft gelatine capsule |
HIV antiviral |
GlaxoSmithKline |
| ALEC |
Dry protein free powder of DPPC-PG |
Liposome |
Lung diseases in infants |
Britannia Pharmaceuticals Ltd |
| Ambisome |
Amphotericin B |
Powder |
Fungal infections |
NeXstar Pharmaceutical Inc |
| Amphocil |
Amphotericin B |
Colloidal dispersion |
Fungal infections |
Sequus Pharmaceutical Inc |
| Amphotec |
Amphotericin B |
Nanoliposome/Solution |
Fungal infections, leishmaniasis |
Sequus Pharmaceutical Inc |
| Aptivus |
tipranavir |
Emulsion/soft gelatine capsule |
AIDS |
Boehringer Ingelheim |
| Atragen |
Tretinoin |
Liposome |
Acute myeloid leukemia |
Aronex Pharmaceuticals Inc |
| Avodart |
Dutasteride |
Emulsion |
Benign Prostatic Hyperplasia |
GSK |
| Avian retrovirus vaccine |
Killed avian retrovirus |
Suspension |
Chickenpox |
Vineland Laboratories |
| Cipro |
Ciprofloxacin |
Oral suspension |
Antibiotic |
Bayer |
| Convulex |
Valproic acid |
Soft gelatine capsule |
Antiepileptic |
Pharmacia |
| DaunoXome |
Daunorubicin citrate |
Solution |
Kaposi sarcoma in AIDS |
NeXstar Pharmaceutical Inc/ Galen Ltd |
| Depakene |
Valproic acid |
Emulsion |
Epilepsy |
Abbott |
| Depocyt |
Cytarabin |
Nanoliposome/Solution |
Lymphomatous meningitis |
Pacira Pharmaceuticals Inc |
| DepoDur |
Morphine |
Suspension |
Post-surgical pain reliever |
Pacira Pharmaceuticals Inc |
| Doxil |
Doxorubicin |
Solution |
Metastatic ovarian, Kaposi sarcoma in AIDS |
Sequus Pharmaceutical Inc |
| Emend |
Aprepitant |
Nanosuspensions/Capsule |
Antiemetic |
Merck-Elan Drug Delivery |
| Epaxal Berna Vaccine |
Inactivated hepatitis-A virions |
Suspension |
Hepatitis A |
Swiss serum & vaccine institute |
| Estrasorb |
estradiol |
Topical emulsion |
Menopausal therapy |
Novavax |
| Evacet |
Doxorubicin |
Liposome |
Metastatic breast cancer |
The liposome company |
| Fenogal |
Fenofibrate |
Tablet |
Anti hyperlipproteinomic |
Genus |
| Fortovase |
saquinavir |
Spontaneously emulsifying systems/soft gelatine capsule |
HIV antiviral |
Roche |
| Fungizone |
Amphotericin B |
Solution |
Fungal infections |
Bristol-Myers Squibb |
| Gengraf |
Cyclosporin A/III |
Spontaneously emulsifying systems/hard gelatine capsule |
Immuno-suppressant |
Abott |
| Hectoral |
Doxercalciferol |
Emulsion |
Calcium regulator |
Bone care |
| Juvela |
Tocopherol nicotinate |
Capsule |
Hypertension, hyperlipidemic |
Eisai Co. |
| Kaletra |
Lopinavir & Ritonavir |
Emulsion/oral solution |
HIV antiviral |
Abott |
| Lamprene |
Clofazamine |
Emulsion |
Leprosy |
Alliance laboratories/ Geigy |
| Lipirex |
fenofibrate |
Hard gelatine capsule |
hyperlipidemia or mixed dyslipidemia |
Sanofi-Aventis |
| Marinol |
Dronabionol |
Emulsion |
Anoxeria |
Roxane |
| Megace ES |
Megestrol acetate |
Nanosuspension |
anorexia, cachexia, weight loss in HIV patients |
Par Pharmaceuticals- Elan Drug Delivery |
| MiKasome |
Amikacin |
Liposome |
Bacterial infection |
NeXstar Pharmaceutical Inc |
| Neoral |
Cyclosporin A/I |
Emulsion |
Immunosuppressant |
Novartis |
| Norvir |
Ritonavir |
Spontaneously emulsifying systems/soft gelatine capsule |
HIV antiviral |
Abott |
| Nyotran |
Nystatin |
Liposome |
Fungal infections |
Aronex pharmaceuticals Inc |
| Panzem NCD |
2-Methoxy estradiol |
Nanosuspension |
anti-proliferative and anti-angiogenic effect |
EntreMed Inc. |
| Prometrium |
Progesterone |
Emulsion |
Endometrial hyperplasia |
Solvay |
| Rapamune |
Sirolimus |
Nanosuspensions/Tablet |
Immunosuppressant |
Wyeth Pharmaceuticals – Elan Drug Delivery |
| Restandol |
Testosterone undecanoate |
Capsules |
Hormone replacement therapy |
Organon laboratories |
| Rocaltrol |
Calcitriol |
Emulsion/soft gelatine capsule |
Calcium regulator |
Roche |
| Sandimmune Neoral |
cyclosporine A/I |
Spontaneously emulsifying systems/soft gelatine capsule |
Immunosuppressant |
Novartis |
| Sustiva |
Efavirenz |
Capsules |
HIV antiviral |
Bristol-Meyers |
| Targretin |
bexarotene |
Soft gelatine capsule |
liver cancer |
Novartis |
| Topex-Br |
Terbutalinesulphate |
Syrup |
Asthma |
Ozone Pharmaceuticals Ltd |
| Tricor |
Fenofibrate |
Nanosuspensions/Tablet |
Antihyperlipidemic agent |
Abbott Laboratories |
| Triglide |
Fenofibrate |
Nanosuspensions/Tablet |
Antihyperlipidemic agent |
Skye Pharma-First Horizon |
| Ventus |
Prostaglandin-E1 |
Liposome |
Systemic inflammatory disease |
The Liposome Company |
| Vesanoid |
tretinoine |
Emulsion/soft gelatine capsule |
Acne |
Roche |
| VincaXome |
Vincristine |
Liposome |
Solid tumors |
NeXstar Pharmaceutical Inc |
| Zemplar |
Paricalcitol |
Emulsion |
Calcium regulator |
Abbott |
LBDDSs formulations to enhance oral delivery of hydrophobic peptide and protein therapeutics
Although there have been promising achievements with LBDDSs for oral delivery of hydrophobic peptide and protein therapeutics, the hydrophilic peptide and protein delivery still remains a challenge and limited to the in-vitro and in-vivo experiments with no product in the pharmaceutical market.
There have been many studies using these lipid-based scaffolds to prove their potential to be exploited in future studies. there have been numerous studies of Insulin as a hydrophilic peptide encapsulated in micelles, microemulsion NPs and nanocapsules with in-situ and in-vivo experiments on rat with promising results as enhanced permeability, bioavailability and efficacy.214-220 In one study SK&F 106760 (a hydrophilic RGD peptide) in the form of microemulsion exploited in in-vivo experiments and demonstrated 50-fold elevated bioavailability.221 In another study Vasopressin was encapsulated as microemulsion in in-situ experiments and resulted in enhanced bioavailability.222 In one study EGF (a single-chain polypeptide) was encapsulated in microemulsions for in-vivo experiments of gastric ulcer in rats and showed increase efficacy.223 In one study on ß-lactamase in-vivo experiments resulted in enhanced 2.5-fold bioavailability.224 N-acetylglucosaminyl and N-acetylmuramyl dipeptide were exploited in one study which demonstrated 10-fold increased bioavailability.225 In another study Leuprolide acetate was encapsulated in microemulsion for in-vivo experiments and proved increased efficacy.226 Two experiments of lipid mixtures with Hexarelin and DMP 728 (Cyclic peptide fibrinogen antagonist) as encapsulated drugs with in-situ experiments showed 20-fold and 3-fold intestinal permeability and bioavailability, respectively.227,228 In the latter study in dog, DuP 532 (an Angiotensin II antagonist) was encapsulated in microemulsion in in-vivo experiments which resulted in 3-fold bioavailability.228 In three studies in rat and pig, calcitonin was encapsulated in mixed micelles and emulsion for in-situ and in-vivo experiments which demonstrated increased permeability and efficacy and 4-fold hypocalcemia response.229-231 Human growth hormone was encapsulated in in-vivo studies on rabbit and showed 3.3% increased bioavailability.232
Regulatory status, commercialization plan and safety information
The status of excipients should be assessed with the regulatory authorities before any pharmaceutical product’s introduction into the market 233 but the expenses of in-vivo toxicity studies are prohibitive for the companies. Such a challenge is happening mainly with the polymeric NPs as there are few of them in the market but lipid NPs owing to their various applications of oils, fats, stabilizer and surfactants have introduced oral and dermal products. The majority of the introduced excipients so far for lipid NPs synthesis are biodegradable, biocompatible and are approved as safe, but some are toxic at high concentrations.234 In this context the FDA has published guide lists of safe materials and substances (GRAS) and inactive ingredient guide (IIG) for excipients that are approved for exploiting in the pharmaceutical products in the market.235 These lists explain and provide insights regarding the appropriate excipient concentration for each administration route and the approved inactive ingredients used for a specific route can be used in all the new formulations. This facilitates the process of synthesizing new formulation as the necessary information can be extracted from the GRAS and IIG. Therefore, such excipients are assessed as substances of a drug not individually as from a “scientific point of view” excipients are a major part of the drug formulation.
Nevertheless, there are other challenges to consider from a “regulatory point of view”; preclinical and clinical studies addressing safety issues and in-vivo manifestations of the LBDDSs in terms of clinical therapeutic efficacy. In-vivo immunological and stability findings toward oils and lipid excipients must be reported to provide in-depth information for the regulatory authorities.236
Besides, factors coming from the biopharmaceuticals are required to be evaluated toward the drug or excipients and this might have paradoxical in-vitro results with in-vivo results due to the physiology of GI tract. Various experiments must be designed and conducted to characterize and recognize the interactions happening among excipients, in-vivo physiological conditions and the drug.237
In order to understand and characterize the in-vivo fate of drugs encapsulated in LBDDSs, a consortium of academic and industrial scientists has been established (http://www.lfcsconsortium.org) which designs experiments to evaluate the function of LBDDSs dispersion and digestion as vital criteria.
Conclusion
Nanotechnology offers promising strategies for enhancing oral bioavailability and therapeutic efficacy of a vast range of drugs; conventional chemical drugs with poor water solubility and biotechnological, peptide and protein therapeutics and biopharmaceuticals. The unique physicochemical features of peptide and protein therapeutics pose challenge for their oral delivery. Hence, their success in site delivery highly depends on technologies and methods to modify these two features not influencing their biological function. In the recent decades numerous DDSs have been introduced and offered by nanotechnology to achieve as high successful delivery as possible and LBDDSs among all has been under investigation owing to their potential for oral delivery of hydrophilic, hydrophobic and lipophilic peptide and protein therapeutics.
LBDDSs enhance solubility and bioavailability of drugs offering strategies such as gastrointestinal lymphatic transport, altering physiological and biochemical properties of gastrointestinal barriers, elevated solubilization and prolonged gastrointestinal retention. Although, such improvements rely on the encapsulation/loading rate and intrinsic composition of the material used during the fabrication process. Obviously, the choice of materials, such as excipients, will influence the success of delivery route which is determined both by lipid formulation design and peptide/protein molecule emphasizing that each peptide/protein-loaded LBDDS must be designed uniquely. Such material must be in correlation with the drug of choice to achieve the maximum therapeutic efficacy and in-site dose.
Most of the scaffolds described in this review article suggest promising alternatives to overcome gastrointestinal enzymatic degradation and poor membrane penetration. Further systematic studies are required to evaluate their in-vivo efficacy in terms of peptide-/protein-based oral drug delivery. Besides “pharmaco-biotechnological” challenges mentioned in this review such as membrane permeability, protease stability, delivery strategies and increased circulation half-life, there are inevitably several “industrial” challenges as well which finally hamper their industrial scale production and consequently their biomedical translation from lab to pharmaceutical market. “Oral bioavailability” still remains the main challenge of peptide/protein-based drug delivery. These factors could be addressed as materials cost, drug potential market feedback, regulatory status, simple industrial-scale fabrication, financial schemes for required instruments, patient compliance administration and high adaptability to human diverse pharmacokinetics.
Competing Interests
The author declares no conflict of interest.
Ethical Approval
There is none to be disclosed.
Funding
The author declares no funding/support.
References
- Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Synthetic therapeutic peptides: science and market. Drug Discov Today 2010; 15(1-2):40-56. doi: 10.1016/j.drudis.2009.10.009 [Crossref] [ Google Scholar]
- Lalatsa A, Schatzlein AG, Uchegbu IF. Strategies to deliver peptide drugs to the brain. Mol Pharm 2014; 11(4):1081-93. doi: 10.1021/mp400680d [Crossref] [ Google Scholar]
- Vajo Z, Fawcett J, Duckworth WC. Recombinant DNA technology in the treatment of diabetes: insulin analogs. Endocr Rev 2001; 22(5):706-17. doi: 10.1210/edrv.22.5.0442 [Crossref] [ Google Scholar]
- Takeda A, Cooper K, Bird A, Baxter L, Frampton GK, Gospodarevskaya E. Recombinant human growth hormone for the treatment of growth disorders in children: a systematic review and economic evaluation. Health Technol Assess 2010; 14(42):1-209. doi: 10.3310/hta14420 [Crossref] [ Google Scholar]
- Cutting GR. Modifier genetics: cystic fibrosis. Annu Rev Genomics Hum Genet 2005; 6:237-60. doi: 10.1146/annurev.genom.6.080604.162254 [Crossref] [ Google Scholar]
- Weatherall DJ. Phenotype-genotype relationships in monogenic disease: lessons from the thalassaemias. Nat Rev Genet 2001; 2(4):245-55. doi: 10.1038/35066048 [Crossref] [ Google Scholar]
- Powell JS. Lasting power of new clotting proteins. Hematology Am Soc Hematol Educ Program 2014; 2014(1):355-63. doi: 10.1182/asheducation-2014.1.355 [Crossref] [ Google Scholar]
- Hirschhorn JN, Lohmueller K, Byrne E, Hirschhorn K. A comprehensive review of genetic association studies. Genet Med 2002; 4(2):45-61. doi: 10.1097/00125817-200203000-00002 [Crossref] [ Google Scholar]
- Savic S, McDermott MF. Clinical genetics in 2014: new monogenic diseases span the immunological disease continuum. Nat Rev Rheumatol 2015; 11(2):67-8. doi: 10.1038/nrrheum.2014.215 [Crossref] [ Google Scholar]
- Hussain N, Jaitley V, Florence AT. Recent advances in the understanding of uptake of microparticulates across the gastrointestinal lymphatics. Adv Drug Deliv Rev 2001; 50(1-2):107-42. doi: 10.1016/s0169-409x(01)00152-1 [Crossref] [ Google Scholar]
- Banga AK. Drug delivery today, Pharma Tech 2002, Business Briefing World Market Series, London, 2002, pp. 150–154.
- Wang W. Instability, stabilization, and formulation of liquid protein pharmaceuticals. Int J Pharm 1999; 185(2):129-88. doi: 10.1016/s0378-5173(99)00152-0 [Crossref] [ Google Scholar]
- Hillery AM, Lloyd AW, Swarbrick J. Drug Delivery and Targeting: For Pharmacists and Pharmaceutical Scientists. 1st ed. London: CRC Press; 2001. 10.1201/b12801.
- Frokjaer S, Otzen DE. Protein drug stability: a formulation challenge. Nat Rev Drug Discov 2005; 4(4):298-306. doi: 10.1038/nrd1695 [Crossref] [ Google Scholar]
- Wang W. Protein aggregation and its inhibition in biopharmaceutics. Int J Pharm 2005; 289(1-2):1-30. doi: 10.1016/j.ijpharm.2004.11.014 [Crossref] [ Google Scholar]
- Rubert Pérez CM, Stephanopoulos N, Sur S, Lee SS, Newcomb C, Stupp SI. The powerful functions of peptide-based bioactive matrices for regenerative medicine. Ann Biomed Eng 2015; 43(3):501-14. doi: 10.1007/s10439-014-1166-6 [Crossref] [ Google Scholar]
- Lax R. The future of peptide development in the pharmaceutical industry. In: PharManufacturing: The International Peptide Review. London, UK: World Business Journals, Pharmaceutical Division; 2010.
- Serrano Lopez DR, Lalatsa A. Peptide pills for brain diseases? Reality and future perspectives. TherDeliv 2013; 4(4):479-501. doi: 10.4155/tde.13.5 [Crossref] [ Google Scholar]
- Adessi C, Soto C. Converting a peptide into a drug: strategies to improve stability and bioavailability. Curr Med Chem 2002; 9(9):963-78. doi: 10.2174/0929867024606731 [Crossref] [ Google Scholar]
- Adessi C, Soto C. Strategies to improve stability and bioavailability of peptide drugs. Front Med Chem 2004; 1(1):513-28. doi: 10.2174/1567204043396622 [Crossref] [ Google Scholar]
- Park K, Kwon IC, Park K. Oral protein delivery: current status and future prospect. React FunctPolym 2011; 71(3):280-7. doi: 10.1016/j.reactfunctpolym.2010.10.002 [Crossref] [ Google Scholar]
- Donovan MD, Flynn GL, Amidon GL. Absorption of polyethylene glycols 600 through 2000: the molecular weight dependence of gastrointestinal and nasal absorption. Pharm Res 1990; 7(8):863-8. doi: 10.1023/a:1015921101465 [Crossref] [ Google Scholar]
- Ikesue K, Kopečkovà P, Kopeček J. Degradation of proteins by guinea pig intestinal enzymes. Int J Pharm 1993; 95(1-3):171-9. doi: 10.1016/0378-5173(93)90404-4 [Crossref] [ Google Scholar]
- Saffran M, Kumar GS, Savariar C, Burnham JC, Williams F, Neckers DC. A new approach to the oral administration of insulin and other peptide drugs. Science 1986; 233(4768):1081-4. doi: 10.1126/science.3526553 [Crossref] [ Google Scholar]
- Fix JA. Oral controlled release technology for peptides: status and future prospects. Pharm Res 1996; 13(12):1760-4. doi: 10.1023/a:1016008419367 [Crossref] [ Google Scholar]
- Lee HJ. Protein drug oral delivery: the recent progress. Arch Pharm Res 2002; 25(5):572-84. doi: 10.1007/bf02976925 [Crossref] [ Google Scholar]
- Lee VH, Satish DK, George MG, Werner R. Oral route of protein and peptide drug delivery. In: Lee VH, ed. Peptide and Protein Drug Delivery. New York: Marcel Dekker; 1991. p. 691-738.
- Pettit DK, Gombotz WR. The development of site-specific drug-delivery systems for protein and peptide biopharmaceuticals. Trends Biotechnol 1998; 16(8):343-9. doi: 10.1016/s0167-7799(98)01186-x [Crossref] [ Google Scholar]
- Tauzin B. Report: Biotechnology Medicines in Development. Washington, DC: Pharmaceutical Research and Manufacturers Association; 2006.
- Crommelin D, van Winden E, Mekking A. Delivery of pharmaceutical proteins. In: Aulton ME, ed. Pharmaceutics: The Science of Dosage form Design. Edinburgh: Churchill Livingstone; 2001. p. 544-53.
- Florence AT, Attwood D. Physicochemical Principles of Pharmacy. London: Pharmaceutical Press; 2006.
- Cleland JL, Langer R. Formulation and delivery of proteins and peptides: design and development strategies. In: Cleland JL, Langer R, eds. Formulation and Delivery of Proteins and Peptides. Washington, DC: American Chemical Society; 1994. p. 1-19.
- Almeida AJ, Souto E. Solid lipid nanoparticles as a drug delivery system for peptides and proteins. Adv Drug Deliv Rev 2007; 59(6):478-90. doi: 10.1016/j.addr.2007.04.007 [Crossref] [ Google Scholar]
- Wang W. Instability, stabilization, and formulation of liquid protein pharmaceuticals. Int J Pharm 1999; 185(2):129-88. doi: 10.1016/s0378-5173(99)00152-0 [Crossref] [ Google Scholar]
- Metselaar JM, Mastrobattista E, Storm G. Liposomes for intravenous drug targeting: design and applications. Mini Rev Med Chem 2002; 2(4):319-29. doi: 10.2174/1389557023405873 [Crossref] [ Google Scholar]
- Gombotz WR, Pettit DK. Biodegradable polymers for protein and peptide drug delivery. Bioconjug Chem 1995; 6(4):332-51. doi: 10.1021/bc00034a002 [Crossref] [ Google Scholar]
- Packhaeuser CB, Schnieders J, Oster CG, Kissel T. In situ forming parenteral drug delivery systems: an overview. Eur J Pharm Biopharm 2004; 58(2):445-55. doi: 10.1016/j.ejpb.2004.03.003 [Crossref] [ Google Scholar]
- Kompella UB, Lee VH. Delivery systems for penetration enhancement of peptide and protein drugs: design considerations. Adv Drug Deliv Rev 2001; 46(1-3):211-45. doi: 10.1016/s0169-409x(00)00137-x [Crossref] [ Google Scholar]
- Prego C, Torres D, Alonso MJ. The potential of chitosan for the oral administration of peptides. Expert Opin Drug Deliv 2005; 2(5):843-54. doi: 10.1517/17425247.2.5.843 [Crossref] [ Google Scholar]
- Ugwoke MI, Agu RU, Verbeke N, Kinget R. Nasal mucoadhesive drug delivery: background, applications, trends and future perspectives. Adv Drug Deliv Rev 2005; 57(11):1640-65. doi: 10.1016/j.addr.2005.07.009 [Crossref] [ Google Scholar]
- Alpar HO, Somavarapu S, Atuah KN, Bramwell VW. Biodegradable mucoadhesive particulates for nasal and pulmonary antigen and DNA delivery. Adv Drug Deliv Rev 2005; 57(3):411-30. doi: 10.1016/j.addr.2004.09.004 [Crossref] [ Google Scholar]
- Schuetz YB, Naik A, Guy RH, Kalia YN. Emerging strategies for the transdermal delivery of peptide and protein drugs. Expert Opin Drug Deliv 2005; 2(3):533-48. doi: 10.1517/17425247.2.3.533 [Crossref] [ Google Scholar]
- Langer R. Where a pill won’t reach. Sci Am 2003; 288(4):50-7. doi: 10.1038/scientificamerican0403-50 [Crossref] [ Google Scholar]
- Myles ME, Neumann DM, Hill JM. Recent progress in ocular drug delivery for posterior segment disease: emphasis on transscleral iontophoresis. Adv Drug Deliv Rev 2005; 57(14):2063-79. doi: 10.1016/j.addr.2005.08.006 [Crossref] [ Google Scholar]
- Smart JD. Buccal drug delivery. Expert Opin Drug Deliv 2005; 2(3):507-17. doi: 10.1517/17425247.2.3.507 [Crossref] [ Google Scholar]
- Crommelin DJ, Storm G, Verrijk R, de Leede L, Jiskoot W, Hennink WE. Shifting paradigms: biopharmaceuticals versus low molecular weight drugs. Int J Pharm 2003; 266(1-2):3-16. doi: 10.1016/s0378-5173(03)00376-4 [Crossref] [ Google Scholar]
- Saltzman WM. Drug Delivery: Engineering Principles for Drug Therapy. New York: Oxford University Press; 2001. 10.1093/oso/9780195085891.003.0005.
- Ugwoke MI, Agu RU, Verbeke N, Kinget R. Nasal mucoadhesive drug delivery: background, applications, trends and future perspectives. Adv Drug Deliv Rev 2005; 57(11):1640-65. doi: 10.1016/j.addr.2005.07.009 [Crossref] [ Google Scholar]
- Myles ME, Neumann DM, Hill JM. Recent progress in ocular drug delivery for posterior segment disease: emphasis on transscleral iontophoresis. Adv Drug Deliv Rev 2005; 57(14):2063-79. doi: 10.1016/j.addr.2005.08.006 [Crossref] [ Google Scholar]
- Smart JD. Buccal drug delivery. Expert Opin Drug Deliv 2005; 2(3):507-17. doi: 10.1517/17425247.2.3.507 [Crossref] [ Google Scholar]
- Mackay M, Phillips J, Hastewell J. Peptide drug delivery: colonic and rectal absorption. Adv Drug Deliv Rev 1997; 28(2):253-73. doi: 10.1016/s0169-409x(97)00076-8 [Crossref] [ Google Scholar]
- Hussain A, Ahsan F. The vagina as a route for systemic drug delivery. J Control Release 2005; 103(2):301-13. doi: 10.1016/j.jconrel.2004.11.034 [Crossref] [ Google Scholar]
- Schuetz YB, Naik A, Guy RH, Kalia YN. Emerging strategies for the transdermal delivery of peptide and protein drugs. Expert Opin Drug Deliv 2005; 2(3):533-48. doi: 10.1517/17425247.2.3.533 [Crossref] [ Google Scholar]
- Agu RU, Ugwoke MI, Armand M, Kinget R, Verbeke N. The lung as a route for systemic delivery of therapeutic proteins and peptides. Respir Res 2001; 2(4):198-209. doi: 10.1186/rr58 [Crossref] [ Google Scholar]
- Bosquillon C, Préat V, Vanbever R. Pulmonary delivery of growth hormone using dry powders and visualization of its local fate in rats. J Control Release 2004; 96(2):233-44. doi: 10.1016/j.jconrel.2004.01.027 [Crossref] [ Google Scholar]
- Ghilzai NM, Desai A. Facing the challenges of transmucosal absorption—buccal, nasal and rectal routes. Pharma Tech 2004, Business Briefing World Market Series, London, 2004, pp. 104–106.
- Choonara BF, Choonara YE, Kumar P, Bijukumar D, du Toit LC, Pillay V. A review of advanced oral drug delivery technologies facilitating the protection and absorption of protein and peptide molecules. Biotechnol Adv 2014; 32(7):1269-82. doi: 10.1016/j.biotechadv.2014.07.006 [Crossref] [ Google Scholar]
- Ensign LM, Cone R, Hanes J. Oral drug delivery with polymeric nanoparticles: the gastrointestinal mucus barriers. Adv Drug Deliv Rev 2012; 64(6):557-70. doi: 10.1016/j.addr.2011.12.009 [Crossref] [ Google Scholar]
- des Rieux A, Fievez V, Garinot M, Schneider YJ, Préat V. Nanoparticles as potential oral delivery systems of proteins and vaccines: a mechanistic approach. J Control Release 2006; 116(1):1-27. doi: 10.1016/j.jconrel.2006.08.013 [Crossref] [ Google Scholar]
- Prego C, Torres D, Fernandez-Megia E, Novoa-Carballal R, Quiñoá E, Alonso MJ. Chitosan-PEG nanocapsules as new carriers for oral peptide delivery Effect of chitosan pegylation degree. J Control Release 2006; 111(3):299-308. doi: 10.1016/j.jconrel.2005.12.015 [Crossref] [ Google Scholar]
- Amidon GL, Lennernäs H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res 1995; 12(3):413-20. doi: 10.1023/a:1016212804288 [Crossref] [ Google Scholar]
- Lipinski CA. Avoiding investment in doomed drugs, is poor solubility an industry wide problem?. Curr Drugs Dis 2001; 4:17-9. [ Google Scholar]
- Murakami T, Takano M. Intestinal efflux transporters and drug absorption. Expert Opin Drug MetabToxicol 2008; 4(7):923-39. doi: 10.1517/17425255.4.7.923 [Crossref] [ Google Scholar]
- Uekama K, Hirayama F, Irie T. Cyclodextrin drug carrier systems. Chem Rev 1998; 98(5):2045-76. doi: 10.1021/cr970025p [Crossref] [ Google Scholar]
- Tauzin B. Report: Biotechnology Medicines in Development. Washington, DC: Pharmaceutical Research and Manufacturers Association; 2019.
- Hillery AM. Drug delivery: the basic concepts. In: Hillery AM, Lloyd AW, Swarbrick J, eds. Drug Delivery and Targeting for Pharmacists and Pharmaceutical Scientists. London: Taylor & Francis; 2001. p. 1-48.
- Frokjaer S, Otzen DE. Protein drug stability: a formulation challenge. Nat Rev Drug Discov 2005; 4(4):298-306. doi: 10.1038/nrd1695 [Crossref] [ Google Scholar]
- Zizzari AT, Pliatsika D, Gall FM, Fischer T, Riedl R. New perspectives in oral peptide delivery. Drug Discov Today 2021; 26(4):1097-1105. doi: 10.1016/j.drudis.2021.01.020 [Crossref] [ Google Scholar]
- Scioli Montoto S, Muraca G, Ruiz ME. Solid Lipid Nanoparticles for Drug Delivery: Pharmacological and Biopharmaceutical Aspects. Front Mol Biosci 2020; 7:587997. doi: 10.3389/fmolb.2020.587997 [Crossref] [ Google Scholar]
- Wang W. Instability, stabilization, and formulation of liquid protein pharmaceuticals. Int J Pharm 1999; 185(2):129-88. doi: 10.1016/s0378-5173(99)00152-0 [Crossref] [ Google Scholar]
- Mehrdadi S. Acute bacterial meningitis: diagnosis, treatment and prevention. J Arch Mil Med 2019; 6(4):e84749. doi: 10.5812/jamm.84749 [Crossref] [ Google Scholar]
- Mehrdadi S. Drug delivery of solid lipid nanoparticles (SLNs) and nanostructured lipid carriers (NLCs) to target brain tumors. Adv Pharm Bull 2023; 13(3):512-20. doi: 10.34172/apb.2023.062 [Crossref] [ Google Scholar]
- Tsomaia N. Peptide therapeutics: targeting the undruggable space. Eur J Med Chem 2015; 94:459-70. doi: 10.1016/j.ejmech.2015.01.014 [Crossref] [ Google Scholar]
- GlobalData. 2015. Available from: http://www.globaldata.com.
- Tomita M, Hayashi M, Horie T, Ishizawa T, Awazu S. Enhancement of colonic drug absorption by the transcellular permeation route. Pharm Res 1988; 5(12):786-9. doi: 10.1023/a:1015992819290 [Crossref] [ Google Scholar]
- Pappenheimer JR. Physiological regulation of transepithelial impedance in the intestinal mucosa of rats and hamsters. J Membr Biol 1987; 100(2):137-48. doi: 10.1007/bf02209146 [Crossref] [ Google Scholar]
- Tang VW, Goodenough DA. Paracellular ion channel at the tight junction. Biophys J 2003; 84(3):1660-73. doi: 10.1016/s0006-3495(03)74975-3 [Crossref] [ Google Scholar]
- Barry PH. Ionic permeation mechanisms in epithelia: biionic potentials, dilution potentials, conductances, and streaming potentials. Methods Enzymol 1989; 171:678-715. doi: 10.1016/s0076-6879(89)71038-7 [Crossref] [ Google Scholar]
- Powell DW. Barrier function of epithelia. Am J Physiol 1981; 241(4):G275-88. doi: 10.1152/ajpgi.1981.241.4.G275 [Crossref] [ Google Scholar]
- Rubas W, Cromwell ME, Shahrokh Z, Villagran J, Nguyen TN, Wellton M. Flux measurements across Caco-2 monolayers may predict transport in human large intestinal tissue. J Pharm Sci 1996; 85(2):165-9. doi: 10.1021/js950267+ [Crossref] [ Google Scholar]
- Madara JL. Regulation of the movement of solutes across tight junctions. Annu Rev Physiol 1998; 60:143-59. doi: 10.1146/annurev.physiol.60.1.143 [Crossref] [ Google Scholar]
- Shakweh M, Ponchel G, Fattal E. Particle uptake by Peyer’s patches: a pathway for drug and vaccine delivery. Expert Opin Drug Deliv 2004; 1(1):141-63. doi: 10.1517/17425247.1.1.141 [Crossref] [ Google Scholar]
- Russell-Jones GJ. Carrier-mediated transport, oral drug delivery. In: Mathiowitz E, ed. Encyclopedia of Controlled Drug Delivery. New York, NY: John Wiley & Sons; 1999. p. 173-84.
- Barthe L, Woodley J, Houin G. Gastrointestinal absorption of drugs: methods and studies. Fundam Clin Pharmacol 1999; 13(2):154-68. doi: 10.1111/j.1472-8206.1999.tb00334.x [Crossref] [ Google Scholar]
- Bai JP, Amidon GL. Structural specificity of mucosal-cell transport and metabolism of peptide drugs: implication for oral peptide drug delivery. Pharm Res 1992; 9(8):969-78. doi: 10.1023/a:1015885823793 [Crossref] [ Google Scholar]
- Russell-Jones GJ. The potential use of receptor-mediated endocytosis for oral drug delivery. Adv Drug Deliv Rev 1996; 20(1):83-97. doi: 10.1016/0169-409x(95)00131-p [Crossref] [ Google Scholar]
- Swaan PW. Recent advances in intestinal macromolecular drug delivery via receptor-mediated transport pathways. Pharm Res 1998; 15(6):826-34. doi: 10.1023/a:1011908128045 [Crossref] [ Google Scholar]
- Charman WN, Porter CJH. Lipophilic prodrugs designed for intestinal lymphatic transport. Adv Drug Deliv Rev 1996; 19(2):149-69. doi: 10.1016/0169-409x(95)00105-g [Crossref] [ Google Scholar]
- Florence AT. Issues in oral nanoparticle drug carrier uptake and targeting. J Drug Target 2004; 12(2):65-70. doi: 10.1080/10611860410001693706 [Crossref] [ Google Scholar]
- Burton PS, Conradi RA, Hilgers AR. (B) Mechanisms of peptide and protein absorption: (2) transcellular mechanism of peptide and protein absorption: passive aspects. Adv Drug Deliv Rev 1991; 7(3):365-85. doi: 10.1016/0169-409x(91)90014-4 [Crossref] [ Google Scholar]
- 91 Giannasca PJ, Giannasca KT, Leichtner AM, Neutra MR. Human intestinal M cells display the sialyl Lewis A antigen. Infect Immun 1999; 67(2):946-53. doi: 10.1128/iai.67.2.946-953.1999 [Crossref] [ Google Scholar]
- Gebert A, Rothkötter HJ, Pabst R. M cells in Peyer’s patches of the intestine. In: Jeon KW, ed. International Review of Cytology. Academic Press; 1996. p. 91-159. 10.1016/s0074-7696(08)61346-7.
- Frey A, Neutra MR. Targeting of mucosal vaccines to Peyer’s patch M cells. Behring Inst Mitt 1997(98):376-89.
- Clark MA, Hirst BH, Jepson MA. Lectin-mediated mucosal delivery of drugs and microparticles. Adv Drug Deliv Rev 2000; 43(2-3):207-23. doi: 10.1016/s0169-409x(00)00070-3 [Crossref] [ Google Scholar]
- Buda A, Sands C, Jepson MA. Use of fluorescence imaging to investigate the structure and function of intestinal M cells. Adv Drug Deliv Rev 2005; 57(1):123-34. doi: 10.1016/j.addr.2004.07.014 [Crossref] [ Google Scholar]
- Jani PU, Florence AT, McCarthy DE. Further histological evidence of the gastrointestinal absorption of polystyrene nanospheres in the rat. Int J Pharm 1992; 84(3):245-52. doi: 10.1016/0378-5173(92)90162-u [Crossref] [ Google Scholar]
- Kataoka K, Tabata J, Yamamoto M, Toyota T. The association of gap junctions with large particles in the crypt epithelium of the rat small intestine. Arch Histol Cytol 1989; 52(2):81-6. doi: 10.1679/aohc.52.81 [Crossref] [ Google Scholar]
- Lavelle EC, Sharif S, Thomas NW, Holland J, Davis SS. The importance of gastrointestinal uptake of particles in the design of oral delivery systems. Adv Drug Deliv Rev 1995; 18(1):5-22. doi: 10.1016/0169-409x(95)00048-c [Crossref] [ Google Scholar]
- O’Hagan DT. Intestinal translocation of particulates—implications for drug and antigen delivery. Adv Drug Deliv Rev 1990; 5(3):265-85. doi: 10.1016/0169-409x(90)90020-s [Crossref] [ Google Scholar]
- Hebden JM, Wilson CG, Spiller RC, Gilchrist PJ, Blackshaw E, Frier ME. Regional differences in quinine absorption from the undisturbed human colon assessed using a timed release delivery system. Pharm Res 1999; 16(7):1087-92. doi: 10.1023/a:1018948102778 [Crossref] [ Google Scholar]
- Shah D, Shen WC. Transcellular delivery of an insulin-transferrin conjugate in enterocyte-like Caco-2 cells. J Pharm Sci 1996; 85(12):1306-11. doi: 10.1021/js9601400 [Crossref] [ Google Scholar]
- Chakraborty S, Shukla D, Mishra B, Singh S. Lipid--an emerging platform for oral delivery of drugs with poor bioavailability. Eur J Pharm Biopharm 2009; 73(1):1-15. doi: 10.1016/j.ejpb.2009.06.001 [Crossref] [ Google Scholar]
- Charman WN, Porter CJ, Mithani S, Dressman JB. Physiochemical and physiological mechanisms for the effects of food on drug absorption: the role of lipids and pH. J Pharm Sci 1997; 86(3):269-82. doi: 10.1021/js960085v [Crossref] [ Google Scholar]
- Crounse RG. Human pharmacology of griseofulvin: the effect of fat intake on gastrointestinal absorption. J Invest Dermatol 1961; 37:529-33. doi: 10.1038/jid.1961.154 [Crossref] [ Google Scholar]
- Hörter D, Dressman JB. Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract. Adv Drug Deliv Rev 2001; 46(1-3):75-87. doi: 10.1016/s0169-409x(00)00130-7 [Crossref] [ Google Scholar]
- Wagner D, Spahn-Langguth H, Hanafy A, Koggel A, Langguth P. Intestinal drug efflux: formulation and food effects. Adv Drug Deliv Rev 2001; 50 Suppl 1:S13-31. doi: 10.1016/s0169-409x(01)00183-1 [Crossref] [ Google Scholar]
- Touitou E, Barry BW. Enhancement in Drug Delivery. Florida: CRC Press; 2006.
- Liversidge GG, Cundy KC. Particle size reduction for improvement of oral bioavailability of hydrophobic drugs: I Absolute oral bioavailability of nanocrystalline danazol in beagle dogs. Int J Pharm 1995; 125(1):91-7. doi: 10.1016/0378-5173(95)00122-y [Crossref] [ Google Scholar]
- Charman WN. Lipids, lipophilic drugs, and oral drug delivery-some emerging concepts. J Pharm Sci 2000; 89(8):967-78. doi: 10.1002/1520-6017(200008)89:8<967::aid-jps1>3.0.co;2-r [Crossref] [ Google Scholar]
- Charman WN, Porter CJ, Mithani S, Dressman JB. Physiochemical and physiological mechanisms for the effects of food on drug absorption: the role of lipids and pH. J Pharm Sci 1997; 86(3):269-82. doi: 10.1021/js960085v [Crossref] [ Google Scholar]
- Porter CJ, Charman WN. Intestinal lymphatic drug transport: an update. Adv Drug Deliv Rev 2001; 50(1-2):61-80. doi: 10.1016/s0169-409x(01)00151-x [Crossref] [ Google Scholar]
- Charman SA, Charman WN, Rogge MC, Wilson TD, Dutko FJ, Pouton CW. Self-emulsifying drug delivery systems: formulation and biopharmaceutic evaluation of an investigational lipophilic compound. Pharm Res 1992; 9(1):87-93. doi: 10.1023/a:1018987928936 [Crossref] [ Google Scholar]
- Li C, Fleisher D, Li L, Schwier JR, Sweetana SA, Vasudevan V. Regional-dependent intestinal absorption and meal composition effects on systemic availability of LY303366, a lipopeptide antifungal agent, in dogs. J Pharm Sci 2001; 90(1):47-57. doi: 10.1002/1520-6017(200101)90:1<47::aidjps6>3.0.co;2-2 [Crossref] [ Google Scholar]
- Martinez M, Amidon G, Clarke L, Jones WW, Mitra A, Riviere J. Applying the biopharmaceutics classification system to veterinary pharmaceutical products Part II Physiological considerations. Adv Drug Deliv Rev 2002; 54(6):825-50. doi: 10.1016/s0169-409x(02)00071-6 [Crossref] [ Google Scholar]
- Sanjula B, Shah FM, Javed A, Alka A. Effect of poloxamer 188 on lymphatic uptake of carvedilol-loaded solid lipid nanoparticles for bioavailability enhancement. J Drug Target 2009; 17(3):249-56. doi: 10.1080/10611860902718672 [Crossref] [ Google Scholar]
- Trevaskis NL, Charman WN, Porter CJ. Lipid-based delivery systems and intestinal lymphatic drug transport: a mechanistic update. Adv Drug Deliv Rev 2008; 60(6):702-16. doi: 10.1016/j.addr.2007.09.007 [Crossref] [ Google Scholar]
- Khoo SM, Shackleford DM, Porter CJ, Edwards GA, Charman WN. Intestinal lymphatic transport of halofantrine occurs after oral administration of a unit-dose lipid-based formulation to fasted dogs. Pharm Res 2003; 20(9):1460-5. doi: 10.1023/a:1025718513246 [Crossref] [ Google Scholar]
- Wang W. Oral protein drug delivery. J Drug Target 1996; 4(4):195-232. doi: 10.3109/10611869608995624 [Crossref] [ Google Scholar]
- Patel G, Misra A. Oral delivery of proteins and peptides: concepts and applications. In: Misra A, ed. Challenges in Delivery of Therapeutic Genomics and Proteomics. London: Elsevier; 2011. p. 481-529. 10.1016/b978-0-12-384964-9.00010-4.
- Woodley JF. Enzymatic barriers for GI peptide and protein delivery. Crit Rev Ther Drug Carrier Syst 1994; 11(2-3):61-95. [ Google Scholar]
- Langguth P, Bohner V, Heizmann J, Merkle HP, Wolffram S, Amidon GL. The challenge of proteolytic enzymes in intestinal peptide delivery. J Control Release 1997; 46(1-2):39-57. doi: 10.1016/s0168-3659(96)01586-6 [Crossref] [ Google Scholar]
- Mrsny R. Challenges for the Oral Delivery of Proteins and Peptides: Theoretical and Practical Approaches to Their Delivery. In: Capsugel Symposia Series. Greenwood, SC; 1991. p. 452.
- Ganong WF. Regulation of gastrointestinal function. In: Ganong WF, ed. Review of Medical Physiology. 12th ed. Lange Medical Publications; 1983. p. 394-420.
- Mrsny R. Challenges for the Oral Delivery of Proteins and Peptides: Theoretical and Practical Approaches to Their Delivery. In: Capsugel Library Symposia series. 1991. p. 45-52.
- Hayakawa E, Lee VH. Aminopeptidase activity in the jejunal and ileal Peyer’s patches of the albino rabbit. Pharm Res 1992; 9(4):535-40. doi: 10.1023/a:1015800615674 [Crossref] [ Google Scholar]
- Skillman JJ, Gould SA, Chung RS, Silen W. The gastric mucosal barrier: clinical and experimental studies in critically ill and normal man, and in the rabbit. Ann Surg 1970; 172(4):564-84. doi: 10.1097/00000658-197010000-00004 [Crossref] [ Google Scholar]
- Meyer RA, McGinley D, Posalaky Z. The gastric mucosal barrier: structure of intercellular junctions in the dog. J Ultrastruct Res 1984; 86(2):192-201. doi: 10.1016/s0022-5320(84)80058-1 [Crossref] [ Google Scholar]
- MacAdam A. The effect of gastro-intestinal mucus on drug absorption. Adv Drug Deliv Rev 1993; 11(3):201-20. doi: 10.1016/0169-409x(93)90010-2 [Crossref] [ Google Scholar]
- Phelps CF. Biosynthesis of mucus glycoprotein. Br Med Bull 1978; 34(1):43-8. doi: 10.1093/oxfordjournals.bmb.a071456 [Crossref] [ Google Scholar]
- Allen A, Garner A. Mucus and bicarbonate secretion in the stomach and their possible role in mucosal protection. Gut 1980; 21(3):249-62. doi: 10.1136/gut.21.3.249 [Crossref] [ Google Scholar]
- Kompella UB, Lee VH. Delivery systems for penetration enhancement of peptide and protein drugs: design considerations. Adv Drug Deliv Rev 2001; 46(1-3):211-45. doi: 10.1016/s0169-409x(00)00137-x [Crossref] [ Google Scholar]
- Kerss S, Allen A, Garner A. A simple method for measuring thickness of the mucus gel layer adherent to rat, frog and human gastric mucosa: influence of feeding, prostaglandin, N-acetylcysteine and other agents. Clin Sci (Lond) 1982; 63(2):187-95. doi: 10.1042/cs0630187 [Crossref] [ Google Scholar]
- Pankhurst QA, Connolly J, Jones SK, Dobson J. Applications of magnetic nanoparticles in biomedicine. J Phys D Appl Phys 2003; 36(13):R167. doi: 10.1088/0022-3727/36/13/201 [Crossref] [ Google Scholar]
- Kreuter J. Nanoparticulate systems in drug delivery and targeting. J Drug Target 1995; 3(3):171-3. doi: 10.3109/10611869509015940 [Crossref] [ Google Scholar]
- Jabir NR, Tabrez S, Ashraf GM, Shakil S, Damanhouri GA, Kamal MA. Nanotechnology-based approaches in anticancer research. Int J Nanomedicine 2012; 7:4391-408. doi: 10.2147/ijn.s33838 [Crossref] [ Google Scholar]
- Li SD, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm 2008; 5(4):496-504. doi: 10.1021/mp800049w [Crossref] [ Google Scholar]
- Pattni BS, Chupin VV, Torchilin VP. New developments in liposomal drug delivery. Chem Rev 2015; 115(19):10938-66. doi: 10.1021/acs.chemrev.5b00046 [Crossref] [ Google Scholar]
- Sawant RR, Torchilin VP. Challenges in development of targeted liposomal therapeutics. AAPS J 2012; 14(2):303-15. doi: 10.1208/s12248-012-9330-0 [Crossref] [ Google Scholar]
- Vieira DB, Gamarra LF. Getting into the brain: liposome-based strategies for effective drug delivery across the blood-brain barrier. Int J Nanomedicine 2016; 11:5381-414. doi: 10.2147/ijn.s117210 [Crossref] [ Google Scholar]
- Craparo EF, Bondì ML, Pitarresi G, Cavallaro G. Nanoparticulate systems for drug delivery and targeting to the central nervous system. CNS NeurosciTher 2011; 17(6):670-7. doi: 10.1111/j.1755-5949.2010.00199.x [Crossref] [ Google Scholar]
- Yang H. Nanoparticle-mediated brain-specific drug delivery, imaging, and diagnosis. Pharm Res 2010; 27(9):1759-71. doi: 10.1007/s11095-010-0141-7 [Crossref] [ Google Scholar]
- Chang HI, Yeh MK. Clinical development of liposome-based drugs: formulation, characterization, and therapeutic efficacy. Int J Nanomedicine 2012; 7:49-60. doi: 10.2147/ijn.s26766 [Crossref] [ Google Scholar]
- Chen W, Li H, Liu Z, Yuan W. Lipopolyplex for therapeutic gene delivery and its application for the treatment of Parkinson’s disease. Front Aging Neurosci 2016; 8:68. doi: 10.3389/fnagi.2016.00068 [Crossref] [ Google Scholar]
- Huang H, Cruz W, Chen J, Zheng G. Learning from biology: synthetic lipoproteins for drug delivery. Wiley Interdiscip Rev NanomedNanobiotechnol 2015; 7(3):298-314. doi: 10.1002/wnan.1308 [Crossref] [ Google Scholar]
- Ag Seleci D, Seleci M, Walter JG, Stahl F, Scheper T. Niosomes as nanoparticular drug carriers: fundamentals and recent applications. J Nanomater 2016; 2016:7372306. doi: 10.1155/2016/7372306 [Crossref] [ Google Scholar]
- Moghassemi S, Hadjizadeh A. Nano-niosomes as nanoscale drug delivery systems: an illustrated review. J Control Release 2014; 185:22-36. doi: 10.1016/j.jconrel.2014.04.015 [Crossref] [ Google Scholar]
- Chaurasia S, Dogra SS. Transfersomes: novel approach for intranasal delivery. Eur J Pharm Med Res 2017; 4(3):192-203. [ Google Scholar]
- Ezzati Nazhad Dolatabadi J, Omidi Y. Solid lipid-based nanocarriers as efficient targeted drug and gene delivery systems. TrAC Trends Anal Chem 2016; 77:100-8. doi: 10.1016/j.trac.2015.12.016 [Crossref] [ Google Scholar]
- Beloqui A, Solinís M, Rodríguez-Gascón A, Almeida AJ, Préat V. Nanostructured lipid carriers: promising drug delivery systems for future clinics. Nanomedicine 2016; 12(1):143-61. doi: 10.1016/j.nano.2015.09.004 [Crossref] [ Google Scholar]
- Eldem T, Speiser P, Hincal A. Optimization of spray-dried and -congealed lipid micropellets and characterization of their surface morphology by scanning electron microscopy. Pharm Res 1991; 8(1):47-54. doi: 10.1023/a:1015874121860 [Crossref] [ Google Scholar]
- Speiser P. LipidnanopelletsalsTrägersystemfürArzneimittelzurperoralenAnwendung. European Patent EP 0167825, 1990.
- Wei L, Yang Y, Shi K, Wu J, Zhao W, Mo J. Preparation and characterization of loperamide-loaded dynasan 114 solid lipid nanoparticles for increased oral absorption in the treatment of diarrhea. Front Pharmacol 2016; 7:332. doi: 10.3389/fphar.2016.00332 [Crossref] [ Google Scholar]
- Subedi RK, Kang KW, Choi HK. Preparation and characterization of solid lipid nanoparticles loaded with doxorubicin. Eur J Pharm Sci 2009; 37(3-4):508-13. doi: 10.1016/j.ejps.2009.04.008 [Crossref] [ Google Scholar]
- Müller RH, Runge SA. Solid lipid nanoparticles (SLN) for controlled drug delivery. In: Benita S, ed. Submicron Emulsions in Drug Targeting and Delivery. Amsterdam: Harwood Academic Publishers; 1998. p. 219-34.
- Üner M, Yener G. Importance of solid lipid nanoparticles (SLN) in various administration routes and future perspectives. Int J Nanomedicine 2007; 2(3):289-300. [ Google Scholar]
- Mukherjee S, Ray S, Thakur RS. Solid lipid nanoparticles: a modern formulation approach in drug delivery system. Indian J Pharm Sci 2009; 71(4):349-58. doi: 10.4103/0250-474x.57282 [Crossref] [ Google Scholar]
- Müller RH, Mäder K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery - a review of the state of the art. Eur J Pharm Biopharm 2000; 50(1):161-77. doi: 10.1016/s0939-6411(00)00087-4 [Crossref] [ Google Scholar]
- Mehnert W, Mäder K. Solid lipid nanoparticles: production, characterization and applications. Adv Drug Deliv Rev 2001; 47(2-3):165-96. doi: 10.1016/s0169-409x(01)00105-3 [Crossref] [ Google Scholar]
- Pandey S, Shaikh F, Gupta A, Tripathi P, Yadav JS. A Recent Update: Solid Lipid Nanoparticles for Effective Drug Delivery. Adv Pharm Bull 2022; 12(1):17-33. doi: 10.34172/apb.2022.007 [Crossref] [ Google Scholar]
- Müller RH, Mehnert W, Lucks J. Solid lipid nanoparticles (SLN)-an alternative colloidal carrier system for controlled drug delivery. Eur J Pharm Biopharm 1995; 41(1):62-9. [ Google Scholar]
- zur Mühlen A, Mehnert W. Drug release and release mechanism of prednisolone loaded solid lipid nanoparticles. Pharmazie 1998; 53(8):552-5. [ Google Scholar]
- Souto EB, Müller RH. Lipid nanoparticles: effect on bioavailability and pharmacokinetic changes. In: Schäfer-Korting M, ed. Drug Delivery. Berlin, Heidelberg: Springer; 2010. p. 115-41. 10.1007/978-3-642-00477-3_4.
- Shah R, Eldridge D, Palombo E, Harding I. Lipid Nanoparticles: Production, Characterization and Stability. New York, NY: Springer International Publishing; 2015.
- Gaba B, Fazil M, Ali A, Baboota S, Sahni JK, Ali J. Nanostructured lipid (NLCs) carriers as a bioavailability enhancement tool for oral administration. Drug Deliv 2015; 22(6):691-700. doi: 10.3109/10717544.2014.898110 [Crossref] [ Google Scholar]
- Shegokar R, Singh KK, Müller RH. Production & stability of stavudine solid lipid nanoparticles--from lab to industrial scale. Int J Pharm 2011; 416(2):461-70. doi: 10.1016/j.ijpharm.2010.08.014 [Crossref] [ Google Scholar]
- Yu YH, Kim E, Park DE, Shim G, Lee S, Kim YB. Cationic solid lipid nanoparticles for co-delivery of paclitaxel and siRNA. Eur J Pharm Biopharm 2012; 80(2):268-73. doi: 10.1016/j.ejpb.2011.11.002 [Crossref] [ Google Scholar]
- Xie S, Zhu L, Dong Z, Wang X, Wang Y, Li X. Preparation, characterization and pharmacokinetics of enrofloxacin-loaded solid lipid nanoparticles: influences of fatty acids. Colloids Surf B Biointerfaces 2011; 83(2):382-7. doi: 10.1016/j.colsurfb.2010.12.014 [Crossref] [ Google Scholar]
- Gastaldi L, Battaglia L, Peira E, Chirio D, Muntoni E, Solazzi I. Solid lipid nanoparticles as vehicles of drugs to the brain: current state of the art. Eur J Pharm Biopharm 2014; 87(3):433-44. doi: 10.1016/j.ejpb.2014.05.004 [Crossref] [ Google Scholar]
- Silva AC, González-Mira E, García ML, Egea MA, Fonseca J, Silva R. Preparation, characterization and biocompatibility studies on risperidone-loaded solid lipid nanoparticles (SLN): high pressure homogenization versus ultrasound. Colloids Surf B Biointerfaces 2011; 86(1):158-65. doi: 10.1016/j.colsurfb.2011.03.035 [Crossref] [ Google Scholar]
- Dwivedi P, Khatik R, Khandelwal K, Taneja I, Raju KS, Wahajuddin Wahajuddin. Pharmacokinetics study of arteether loaded solid lipid nanoparticles: an improved oral bioavailability in rats. Int J Pharm 2014; 466(1-2):321-7. doi: 10.1016/j.ijpharm.2014.03.036 [Crossref] [ Google Scholar]
- Raza K, Singh B, Singal P, Wadhwa S, Katare OP. Systematically optimized biocompatible isotretinoin-loaded solid lipid nanoparticles (SLNs) for topical treatment of acne. Colloids Surf B Biointerfaces 2013; 105:67-74. doi: 10.1016/j.colsurfb.2012.12.043 [Crossref] [ Google Scholar]
- Ravi PR, Vats R, Dalal V, Murthy AN. A hybrid design to optimize preparation of lopinavir loaded solid lipid nanoparticles and comparative pharmacokinetic evaluation with marketed lopinavir/ritonavir coformulation. J Pharm Pharmacol 2014; 66(7):912-26. doi: 10.1111/jphp.12217 [Crossref] [ Google Scholar]
- Kheradmandnia S, Vasheghani-Farahani E, Nosrati M, Atyabi F. Preparation and characterization of ketoprofen-loaded solid lipid nanoparticles made from beeswax and carnauba wax. Nanomedicine 2010; 6(6):753-9. doi: 10.1016/j.nano.2010.06.003 [Crossref] [ Google Scholar]
- Madan J, Pandey RS, Jain V, Katare OP, Chandra R, Katyal A. Poly (ethylene)-glycol conjugated solid lipid nanoparticles of noscapine improve biological half-life, brain delivery and efficacy in glioblastoma cells. Nanomedicine 2013; 9(4):492-503. doi: 10.1016/j.nano.2012.10.003 [Crossref] [ Google Scholar]
- Mehnert W, Mäder K. Solid lipid nanoparticles: production, characterization and applications. Adv Drug Deliv Rev 2001; 47(2-3):165-96. doi: 10.1016/s0169-409x(01)00105-3 [Crossref] [ Google Scholar]
- Wang S, Chen T, Chen R, Hu Y, Chen M, Wang Y. Emodin loaded solid lipid nanoparticles: preparation, characterization and antitumor activity studies. Int J Pharm 2012; 430(1-2):238-46. doi: 10.1016/j.ijpharm.2012.03.027 [Crossref] [ Google Scholar]
- Kuo YC, Wang CC. Cationic solid lipid nanoparticles with primary and quaternary amines for release of saquinavir and biocompatibility with endothelia. Colloids Surf B Biointerfaces 2013; 101:101-5. doi: 10.1016/j.colsurfb.2012.06.002 [Crossref] [ Google Scholar]
- Cai S, Yang Q, Bagby TR, Forrest ML. Lymphatic drug delivery using engineered liposomes and solid lipid nanoparticles. Adv Drug Deliv Rev 2011; 63(10-11):901-8. doi: 10.1016/j.addr.2011.05.017 [Crossref] [ Google Scholar]
- Nafee N, Husari A, Maurer CK, Lu C, de Rossi C, Steinbach A. Antibiotic-free nanotherapeutics: ultra-small, mucus-penetrating solid lipid nanoparticles enhance the pulmonary delivery and anti-virulence efficacy of novel quorum sensing inhibitors. J Control Release 2014; 192:131-40. doi: 10.1016/j.jconrel.2014.06.055 [Crossref] [ Google Scholar]
- Almeida AJ, Souto E. Solid lipid nanoparticles as a drug delivery system for peptides and proteins. Adv Drug Deliv Rev 2007; 59(6):478-90. doi: 10.1016/j.addr.2007.04.007 [Crossref] [ Google Scholar]
- Shah RM, Malherbe F, Eldridge D, Palombo EA, Harding IH. Physicochemical characterization of solid lipid nanoparticles (SLNs) prepared by a novel microemulsion technique. J Colloid Interface Sci 2014; 428:286-94. doi: 10.1016/j.jcis.2014.04.057 [Crossref] [ Google Scholar]
- Soares S, Fonte P, Costa A, Andrade J, Seabra V, Ferreira D. Effect of freeze-drying, cryoprotectants and storage conditions on the stability of secondary structure of insulin-loaded solid lipid nanoparticles. Int J Pharm 2013; 456(2):370-81. doi: 10.1016/j.ijpharm.2013.08.076 [Crossref] [ Google Scholar]
- Kalhapure RS, Mocktar C, Sikwal DR, Sonawane SJ, Kathiravan MK, Skelton A. Ion pairing with linoleic acid simultaneously enhances encapsulation efficiency and antibacterial activity of vancomycin in solid lipid nanoparticles. Colloids Surf B Biointerfaces 2014; 117:303-11. doi: 10.1016/j.colsurfb.2014.02.045 [Crossref] [ Google Scholar]
- Hao J, Wang F, Wang X, Zhang D, Bi Y, Gao Y. Development and optimization of baicalin-loaded solid lipid nanoparticles prepared by coacervation method using central composite design. Eur J Pharm Sci 2012; 47(2):497-505. doi: 10.1016/j.ejps.2012.07.006 [Crossref] [ Google Scholar]
- Pardeshi C, Rajput P, Belgamwar V, Tekade A, Patil G, Chaudhary K. [Solid lipid based nanocarriers: an overview]. Acta Pharm 2013; 62(4):433-72. doi: 10.2478/v10007-012-0040-z [Crossref] [ Google Scholar]
- Muchow M, Maincent P, Muller RH. Lipid nanoparticles with a solid matrix (SLN, NLC, LDC) for oral drug delivery. Drug Dev Ind Pharm 2008; 34(12):1394-405. doi: 10.1080/03639040802130061 [Crossref] [ Google Scholar]
- Yun Y, Cho YW, Park K. Nanoparticles for oral delivery: targeted nanoparticles with peptidic ligands for oral protein delivery. Adv Drug Deliv Rev 2013; 65(6):822-832. doi: 10.1016/j.addr.2012.10.007 [Crossref] [ Google Scholar]
- Borgström B. Importance of phospholipids, pancreatic phospholipase A2, and fatty acid for the digestion of dietary fat: in vitro experiments with the porcine enzymes. Gastroenterology 1980; 78(5 Pt 1):954-62. [ Google Scholar]
- Borgström B, Donnér J. The polar interactions between pancreatic lipase, colipase and the triglyceride substrate. FEBS Lett 1977; 83(1):23-6. doi: 10.1016/0014-5793(77)80633-9 [Crossref] [ Google Scholar]
- Scow RO, Olivecrona T. Effect of albumin on products formed from chylomicron triacylclycerol by lipoprotein lipase in vitro. BiochimBiophys Acta 1977; 487(3):472-86. doi: 10.1016/0005-2760(77)90217-x [Crossref] [ Google Scholar]
- Olbrich C, Müller RH. Enzymatic degradation of SLN-effect of surfactant and surfactant mixtures. Int J Pharm 1999; 180(1):31-9. doi: 10.1016/s0378-5173(98)00404-9 [Crossref] [ Google Scholar]
- Müller RH, Olbrich C. Solid lipid nanoparticles: phagocytic uptake, in vitro cytotoxicity and in vitro biodegradation, 2nd communication. Pharm Ind 1999; 61(6):564-9. [ Google Scholar]
- Olbrich C, Mehnert W, Müller RH. In vitro degradation properties of solid lipid nanoparticles. in: Proc. 2nd World Meeting APGI/APV; Paris; 1998. p. 577-8.
- Müller RH, Rühl D, Runge SA. Biodegradation of solid lipid nanoparticles as a function of lipase incubation time. Int J Pharm 1996; 144(1):115-21. doi: 10.1016/s0378-5173(96)04731-x [Crossref] [ Google Scholar]
- Olbrich C, Mehnert W, Müller RH. In vitro degradation properties of solid lipid nanoparticles. in: Proc. 2nd World Meeting APGI/APV; Paris; 1998. p. 627-8.
- Yang S, Zhu J, Lu Y, Liang B, Yang C. Body distribution of camptothecin solid lipid nanoparticles after oral administration. Pharm Res 1999; 16(5):751-7. doi: 10.1023/a:1018888927852 [Crossref] [ Google Scholar]
- Penkler L, Müller RH, Runge SA. Pharmaceutical Cyclosporin Formulation with Improved Biopharmaceutical Properties, Improved Physical Quality and Greater Stability, and Method for Producing Said Formulation. WO 99/56733, 1999.
- Bargoni A, Cavalli R, Caputo O, Fundarò A, Gasco MR, Zara GP. Solid lipid nanoparticles in lymph and plasma after duodenal administration to rats. Pharm Res 1998; 15(5):745-50. doi: 10.1023/a:1011975120776 [Crossref] [ Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983; 65(1-2):55-63. doi: 10.1016/0022-1759(83)90303-4 [Crossref] [ Google Scholar]
- Müller RH, Maaβen S, Weyhers H, Specht F, Lucks JS. Cytotoxicity of magnetite-loaded polylactide, polylactide/glycolide particles and solid lipid nanoparticles. Int J Pharm 1996; 138(1):85-94. doi: 10.1016/0378-5173(96)04539-5 [Crossref] [ Google Scholar]
- Marcato PD, Durán N. Cytotoxicity and genotoxicity of solid lipid nanoparticles. In: Durán N, Guterres SS, Alves OL, eds. Nanotoxicology: Materials, Methodologies, and Assessments. New York, NY: Springer; 2014. p. 229-44. 10.1007/978-1-4614-8993-1_10.
- Almeida H, Amaral MH, Lobão P, Silva AC, Loboa JM. Applications of polymeric and lipid nanoparticles in ophthalmic pharmaceutical formulations: present and future considerations. J Pharm Pharm Sci 2014; 17(3):278-93. doi: 10.18433/j3dp43 [Crossref] [ Google Scholar]
- Sandeep Kalepu, Mohanvarma Manthina, Veerabhadhraswamy Padavala. Oral lipid-based drug delivery systems – an overview. Acta Pharmaceutica Sinica B 2013; 3(6):361-372. doi: 10.1016/j.apsb.2013.10.001 [Crossref] [ Google Scholar]
- Doktorovova S, Souto EB, Silva AM. Nanotoxicology applied to solid lipid nanoparticles and nanostructured lipid carriers - a systematic review of in vitro data. Eur J Pharm Biopharm 2014; 87(1):1-18. doi: 10.1016/j.ejpb.2014.02.005 [Crossref] [ Google Scholar]
- Fortunov S, Peneva P, Andonova V, Kassarova M. Toxicity assessment of drug delivery nanocarriers. Sci Technol 2016; 6(1):262-6. [ Google Scholar]
- Hillegass JM, Shukla A, Lathrop SA, MacPherson MB, Fukagawa NK, Mossman BT. Assessing nanotoxicity in cells in vitro. Wiley Interdiscip Rev NanomedNanobiotechnol 2010; 2(3):219-31. doi: 10.1002/wnan.54 [Crossref] [ Google Scholar]
- Maaßen S, Schwarz C, Mehnert W, Lucks JS, Yunis-Specht F, Muller BW. Comparison of cytotoxicity between polyester nanoparticles and solid lipid nanoparticles. Proc Int Symp Control Rel Bioact Mater 1993; 20:490-1. [ Google Scholar]
- Müller RH, Maassen S, Weyhers H, Mehnert W. Phagocytic uptake and cytotoxicity of solid lipid nanoparticles (SLN) sterically stabilized with poloxamine 908 and poloxamer 407. J Drug Target 1996; 4(3):161-70. doi: 10.3109/10611869609015973 [Crossref] [ Google Scholar]
- Yu YH, Kim E, Park DE, Shim G, Lee S, Kim YB. Cationic solid lipid nanoparticles for co-delivery of paclitaxel and siRNA. Eur J Pharm Biopharm 2012; 80(2):268-73. doi: 10.1016/j.ejpb.2011.11.002 [Crossref] [ Google Scholar]
- Shi S, Han L, Deng L, Zhang Y, Shen H, Gong T. Dual drugs (microRNA-34a and paclitaxel)-loaded functional solid lipid nanoparticles for synergistic cancer cell suppression. J Control Release 2014; 194:228-37. doi: 10.1016/j.jconrel.2014.09.005 [Crossref] [ Google Scholar]
- Jin J, Bae KH, Yang H, Lee SJ, Kim H, Kim Y. In vivo specific delivery of c-Met siRNA to glioblastoma using cationic solid lipid nanoparticles. Bioconjug Chem 2011; 22(12):2568-72. doi: 10.1021/bc200406n [Crossref] [ Google Scholar]
- Baek JS, Cho CW. Controlled release and reversal of multidrug resistance by co-encapsulation of paclitaxel and verapamil in solid lipid nanoparticles. Int J Pharm 2015; 478(2):617-24. doi: 10.1016/j.ijpharm.2014.12.018 [Crossref] [ Google Scholar]
- Kalhapure RS, Mocktar C, Sikwal DR, Sonawane SJ, Kathiravan MK, Skelton A. Ion pairing with linoleic acid simultaneously enhances encapsulation efficiency and antibacterial activity of vancomycin in solid lipid nanoparticles. Colloids Surf B Biointerfaces 2014; 117:303-11. doi: 10.1016/j.colsurfb.2014.02.045 [Crossref] [ Google Scholar]
- Morishita M, Morishita I, Takayama K, Machida Y, Nagai T. Site-dependent effect of aprotinin, sodium caprate, Na2EDTA and sodium glycocholate on intestinal absorption of insulin. Biol Pharm Bull 1993; 16(1):68-72. doi: 10.1248/bpb.16.68 [Crossref] [ Google Scholar]
- Lane ME, O’Driscoll C M, Corrigan OI. Quantitative estimation of the effects of bile salt surfactant systems on insulin stability and permeability in the rat intestine using a mass balance model. J Pharm Pharmacol 2005; 57(2):169-75. doi: 10.1211/0022357055434 [Crossref] [ Google Scholar]
- Sharma G, Wilson K, van der Walle CF, Sattar N, Petrie JR, Ravi Kumar MN. Microemulsions for oral delivery of insulin: design, development and evaluation in streptozotocin induced diabetic rats. Eur J Pharm Biopharm 2010; 76(2):159-69. doi: 10.1016/j.ejpb.2010.07.002 [Crossref] [ Google Scholar]
- Cilek A, Celebi N, Tirnaksiz F, Tay A. A lecithin-based microemulsion of rh-insulin with aprotinin for oral administration: Investigation of hypoglycemic effects in non-diabetic and STZ-induced diabetic rats. Int J Pharm 2005; 298(1):176-85. doi: 10.1016/j.ijpharm.2005.04.016 [Crossref] [ Google Scholar]
- Elsayed A, Remawi MA, Qinna N, Farouk A, Badwan A. Formulation and characterization of an oily-based system for oral delivery of insulin. Eur J Pharm Biopharm 2009; 73(2):269-79. doi: 10.1016/j.ejpb.2009.06.004 [Crossref] [ Google Scholar]
- Toorisaka E, Ono H, Arimori K, Kamiya N, Goto M. Hypoglycemic effect of surfactant-coated insulin solubilized in a novel solid-in-oil-in-water (S/O/W) emulsion. Int J Pharm 2003; 252(1-2):271-4. doi: 10.1016/s0378-5173(02)00674-9 [Crossref] [ Google Scholar]
- Watnasirichaikul S, Rades T, Tucker IG, Davies NM. In-vitro release and oral bioactivity of insulin in diabetic rats using nanocapsules dispersed in biocompatible microemulsion. J Pharm Pharmacol 2002; 54(4):473-80. doi: 10.1211/0022357021778736 [Crossref] [ Google Scholar]
- Constantinides PP, Scalart JP, Lancaster C, Marcello J, Marks G, Ellens H. Formulation and intestinal absorption enhancement evaluation of water-in-oil microemulsions incorporating medium-chain glycerides. Pharm Res 1994; 11(10):1385-90. doi: 10.1023/a:1018927402875 [Crossref] [ Google Scholar]
- Ritschel WA. Microemulsions for improved peptide absorption from the gastrointestinal tract. Methods Find Exp Clin Pharmacol 1991; 13(3):205-20. [ Google Scholar]
- Celebi N, Türkyilmaz A, Gönül B, Ozogul C. Effects of epidermal growth factor microemulsion formulation on the healing of stress-induced gastric ulcers in rats. J Control Release 2002; 83(2):197-210. doi: 10.1016/s0168-3659(02)00198-0 [Crossref] [ Google Scholar]
- Rao SV, Yajurvedi K, Shao J. Self-nanoemulsifying drug delivery system (SNEDDS) for oral delivery of protein drugs: III In vivo oral absorption study. Int J Pharm 2008; 362(1-2):16-9. doi: 10.1016/j.ijpharm.2008.05.015 [Crossref] [ Google Scholar]
- Lyons KC, Charman WN, Miller R, Porter CJ. Factors limiting the oral bioavailability of N-acetylglucosaminyl-N-acetylmuramyl dipeptide (GMDP) and enhancement of absorption in rats by delivery in a water-in-oil microemulsion. Int J Pharm 2000; 199(1):17-28. doi: 10.1016/s0378-5173(00)00349-5 [Crossref] [ Google Scholar]
- Zheng JY, Fulu MY. Decrease of genital organ weights and plasma testosterone levels in rats following oral administration of leuprolide microemulsion. Int J Pharm 2006; 307(2):209-15. doi: 10.1016/j.ijpharm.2005.10.007 [Crossref] [ Google Scholar]
- Fagerholm U, Sjöström B, Sroka-Markovic J, Wijk A, Svensson M, Lennernäs H. The effect of a drug-delivery system consisting of soybean phosphatidyl choline and medium-chain monoacylglycerol on the intestinal permeability of hexarelin in the rat. J Pharm Pharmacol 1998; 50(5):467-73. doi: 10.1111/j.2042-7158.1998.tb06187.x [Crossref] [ Google Scholar]
- Aungst BJ, Saitoh H, Burcham DL, Huang SM, Mousa SA, Hussain MA. Enhancement of the intestinal absorption of peptides and nonpeptides. J Control Release 1996; 41(1):19-31. doi: 10.1016/0168-3659(96)01353-3 [Crossref] [ Google Scholar]
- Hastewell J, Lynch S, Williamson I, Fox R, Mackay M. Absorption of human calcitonin across the rat colon in vivo. Clin Sci (Lond) 1992; 82(5):589-94. doi: 10.1042/cs0820589 [Crossref] [ Google Scholar]
- New R, Littlewood G, Guard P, Browning I, Hotten P. Intestinal delivery of calcitonin in pig. Int J Pharm 1997; 156(1):1-8. doi: 10.1016/s0378-5173(97)00130-0 [Crossref] [ Google Scholar]
- Fan Y, Li X, Zhou Y, Fan C, Wang X, Huang Y. Improved intestinal delivery of salmon calcitonin by water-in-oil microemulsions. Int J Pharm 2011; 416(1):323-30. doi: 10.1016/j.ijpharm.2011.06.029 [Crossref] [ Google Scholar]
- Yoshiura H, Tahara Y, Hashida M, Kamiya N, Hirata A, Fujii T. Design and in vivo evaluation of solid-in-oil suspension for oral delivery of human growth hormone. BiochemEng J 2008; 41(2):106-10. doi: 10.1016/j.bej.2008.04.001 [Crossref] [ Google Scholar]
- FDA. 21 CFR Part 182, Substances Generally Recognized as Safe. FDA; 2010.
- U.S. Department of Health and Human Services, Food and Drug Administration. Guidance for Industry: Nonclinical Studies for the Safety Evaluation of Pharmaceutical Excipients. Office of Training and Communication, Division of Drug Information, HFD-240, Center for Drug Evaluation and Research, Food and Drug Administration, or Office of Communication, Training, and Manufacturers Assistance, HFM-40, Center for Biologics Evaluation and Research, Food and Drug Administration. May 2005. Available from: http://www.fda.gov/cder/guidance/5544fnl.pdf.
- U.S. Food and Drug Administration. Inactive Ingredients Search for Approved Drug Products. Division of Labeling and Program Support, Office of Generic Drugs, Center for Drug Evaluation and Research, Food and Drug Administration; 2007. Available from: http://www.accessdata.fda.gov/scripts/cder/iig/index.cfm.
- Maincent P. The regulatory environment: the challenges for lipid-based formulation. Bull Tech Gattefossé 2007; 100:47-9. [ Google Scholar]
- Chen ML. Lipid excipients and delivery systems for pharmaceutical development: a regulatory perspective. Adv Drug Deliv Rev 2008; 60(6):768-77. doi: 10.1016/j.addr.2007.09.010 [Crossref] [ Google Scholar]